
Symmetrical 1-pyrrolidineacetamide showing anti-HIV activity through a new binding site on HIV-1 integrase1
Introduction
An essential step in the HIV-1 life cycle is the integration of the reverse-transcribed viral genome into host chromosomal DNA by the virally-encoded integrase (IN) protein[1]. The fact that its inactivation either by mutagenesis or inhibition might block the productive infection by HIV-1[2,3] implies that HIV-1 IN is an attractive target for antiviral drug discovery. IN catalyzes 2 steps of reaction of the integration process. In the first step, termed 3´-processing, IN cleaves the 2 terminal nucleotides from each 3´ end of the viral DNA. The second step is called strand transfer, in which IN transfers both extremities of the viral DNA into the target DNA with the help of some cellular enzymes by a 1-step transesterification reaction, resulting in full-site integration[4–6].
HIV-1 IN is composed of 3 distinct structural and functional domains: the N-terminal domain (residues 1−50) that contains a conserved HHCC zinc-binding motif, the core domain (residues 51−212) that contains the catalytic site with 3 spatially conserved and invariable amino acids (D64, D116, and E152), and the C-terminal domain (residues 213−270), which is suggested to be responsible to multimer formation and non-specific binding to DNA[7,8]. To date, crystallographic or Nuclear Magnetic Resonance (NMR) structural data have been available for each of the IN individual domains, and 2-domain crystal structures (either the core and C-terminal domains or N-terminal and the core domains) have been determined[9,10]. However, in the 2-domain integrase structures, the positioning of both the N- and C-terminal domains in relation to the catalytic core domain (CCD) may not correspond to that assumed when viral DNA is bound. Efforts to obtain a structure of the full-length IN have been impeded by poor protein solubility.
The structural and biochemical understanding of IN has led to the discovery and development of diverse classes of active compounds against IN. The most promising drug candidate is of β-diketo type, which is the only class of IN inhibitors with a clear inhibition mechanism[11,12]. Among the group, S-1360 and L-870, 810 entered phase II clinical trials in 2003 and 2004[7,13] and failed later. However, although various kinds of IN inhibitors have been reported, and some have been used in clinical trials, only 1 IN inhibitor, raltegravir, was approved by the Food and Drug Administration (FDA) in 2007[7,11,13,14]. Therefore, the discovery of a novel IN potent inhibitor is still an alluring project.
It is well known that the structural information detailing the association between IN and potential inhibitors is of highly-therapeutic importance. Identification of the key amino acid residues involved in the binding site of candidate drugs would help to predict drug-resistant viral strains and provide specific information for inhibitor modification[15–17].
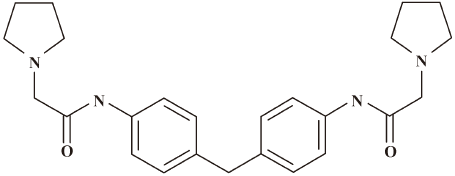
In this work, we report a small molecular compound (compound 1; Figure 1) 1-pyrrolidineacetamide, N,N´-(methylene-di-4,1-phenylene)bis-1-pyrrolidineacetamide that could competitively inhibit IN binding to viral DNA and show moderate antiretroviral activity. Site-directed mutagenesis with molecular docking analyses revealed that compound 1 binds to IN with key residues at the CCD dimer interface. Our current study is expected to provide some useful information for the discovery of IN-based anti-HIV inhibitors.
Materials and methods
Chemistry Compound 1 was purchased from SPECS Bank (Delft, Netherlands).
Plasmid construction The wild-type HIV-1 IN DNA coding for HIV-1 integrase (GenBank N
Protein preparation The proteins IN, IN52–210, and IN mutants K103A, K173A, and T174A were expressed and purified according to the GST Gene Fusion System Handbook (Amersham Bioscience, Pittsburgh, PA, USA). In brief, Escherichia coli BL21(DE3) cells transformed with wild-type or mutated HIV-1 IN expression plasmids were grown at 37 °C in Lysogeny Broth (LB) medium containing 100 µg/mL ampicillin until the optical density at 600 nm reached 0.6−0.8. Proteins were expressed for 5−8 h at 25 °C after induction with 0.5 mmol/L isopropyl-β-D-thiogalactopyranoside (IPTG). The cells were harvested by centrifugation, resuspended in 1×precooled phosphate-buffered saline (PBS; 140 mmol/L NaCl, 2.7 mmol/L KCl, 10 mmol/L Na2HPO4, and 1.8 mmol/L KH2PO4, pH 7.4), and lysed by sonication in an ice bath. The lysate was centrifuged for 30 min at 21 000×g at 4 °C. The supernatant was loaded on a glutathione–Sepharose 4B column (Amersham–Pharmacia, Pittsburgh, PA, USA) equilibrated with PBS at 4 °C. The column was washed with 120−200 mL of 1× PBS and then eluted with 10 mL of 20 mmol/L reduced glutathione. The elution fraction was applied on a Superdex 75 column on an AKTA instrument (Amersham–Pharmacia, Pittsburgh, PA, USA) for further purification. IN and IN mutants K103A, K173A, and T174A were purified in GST fusion form. For IN52–210, the GST-fusion protein was digested on the glutathione–Sepharose 4B column with 50 U thrombin for 16 h at 4 °C to remove the GST fusion tag. The purity of all proteins was confirmed by SDS–PAGE.
Competitive inhibition assay The compound’s competitive inhibition assay against IN/viral DNA binding was performed by using the surface plasma resonance (SPR) biosensor technology-based Biacore 3000 system (Biacore AB, Uppsala, Sweden) as previously reported[19]. During the assay, a 21 bp 5´-biotinylated oligonucleotide (5´-GTGTGGAAAATCTCTAGGTGT-3´) hybridized with a non-biotinylated complementary oligonucleotide was immobilized on the streptavidin matrix-coated sensor chip (SA chip), and 200 nmol/L IN incubated with 0–0.2 mmol/L compounds for 1 h at 4 °C flowed over the chip surface. A 21 bp DNA with random sequence was immobilized to the reference flow cell as the control. The compound inhibition against IN binding to DNA was demonstrated by monitoring the response unit (RU) decrease with the addition of the compounds at different concentrations. All the sensorgrams were processed by using automatic correction for non-specific bulk refractive index effects.
Binding assay The binding affinity of the compound to HIV-1 IN, IN52–210, IN(K103A), IN(K173A), and IN(T174A) in vitro was determined by using SPR technology. The measurement was performed using the dual flow cell Biacore 3000 instrument. Immobilization of the wild-type and mutant IN proteins to the hydrophilic carboxymethylated dextran matrix of the sensor chip CM5 (Biacore, Sweden) was carried out by the standard primary amine coupling method. The protein to be covalently bound to the matrix was diluted in 10 mmol/L sodium acetate buffer (pH 4.5) to a final concentration of 0.2 mg/mL, and the resonance signal reached approximately 8500 RU. Equilibration of the baseline was completed by a continuous flow of HBS–EP buffer (10 mmol/L HEPES, 150 mmol/L NaCl, 3 mmol/L EDTA and 0.01% P20, pH 7.4) through the chip for 4−5 h. For the GST fusion protein, IN, IN(K103A), IN(K173A), and IN(T174A) binding assays, the reference flow cell surface was immobilized at a parallel level (4500 RU) using GST as a control. All the sensorgrams were processed by using automatic correction for non-specific bulk refractive index effects. The specific binding profiles of the compounds to the immobilized protein were obtained after subtracting the response signal from the control flow cell. All the Biacore data were collected at 25 °C with HBS–EP as the running buffer at a constant flow of 30 µL/min. The equilibrium dissociation constants (KD) evaluating the protein–ligand binding affinity were determined using the 1:1 binding model (Langmuir), and the curve fitting efficiency was checked by residual plots and χ2.
Antiretroviral activity assay The C8166 cells were grown and maintained in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum, 2 mmol/L L-glutamine, 0.1% sodium bicarbonate, and 20 μg gentamicin per mL. HIV-1(IIIB) was obtained from Medical Research Council, AIDS Reagent Project (London, UK). The inhibitory effect of the compound on HIV-1 replication was monitored by the inhibition of virus-induced cytopathicity in the C8166 cells for 5 d after infection as described. Cytotoxicity of the compounds against the C8166 cells was determined by measuring the viability after 5 d of incubation[20–22].
Molecular modeling The computational molecular modeling studies were carried out using a Dell Precision 670 workstation (Austin, TX, USA) running Redhat Linux WS 3.0 (Redhat, Raleigh, NC, USA). The 3-D structure of compound 1 was constructed and energetically minimized with Gasteiger–Hückel charges[23] and the Tripos force field[24] in molecular modeling software package SYBYL version 7.0[25]. Both the nitrogen atoms in the pyrrolidines were protonated, which is consistent with the condition of the binding assay (pH 7.4). The 3-D crystal structure of HIV-1 IN CCD was obtained from the Protein Data Bank[26] (PDB) with entry code 1QS4[27]. Only chains A B of 1QS4 were used in this study. After all of the hydrogen atoms were added, the Glu131 residues in the surface of the CCD dimer were mutated back to Trp. The hydrogen atoms and the mutated Glu131 residues were then minimized using the Kollman all-atom force field[28] with Kollman all-atom charges[29] in SYBYL.
GOLD version 3.0.1 (Cambridge Crystallographic Data Centre, Cambridge, UK) was used to investigate the reasonable binding site[30]. In the docking, the ligand was flexible, and the protein remained rigid. All the protonation of the histine residues were maintained as in crystal structure without special treatment. The whole CCD dimer was treated as the binding pocket by defining 2 active center atoms with a 30 Å radius first, respectively. Then only the potential binding site of the CCD was included in the following docking. During the GOLD docking, the default parameters of genetic algorithms were applied to search the reasonable binding conformation of compound 1. To ensure the atoms’ type, both ligand and protein were turned on in the option “set atom type”. To find more accurate geometries, the option “allow early termination” was turned off. The GOLDScore function was used to evaluate the docking results[31,32].
Results
Compound 1 could inhibit HIV-1 integrase binding to viral DNA as a catalytic core domain binder Com-pound 1 was identified from other compounds by random screening against HIV-1 IN. The SPR technology-based competitive inhibition assay revealed that compound 1 could compete the binding of IN to the immobilized viral DNA in a dose-dependent manner. As shown in Figure 2A, the RU values of IN binding to the viral DNA significantly decreased with the increase of compound concentrations. The IC50 value for compound 1 was therefore evaluated as 7.29±0.68 µmol/L (Figure 2B) by fitting the inhibition data to a dose-dependent curve using a logistic derivative equation (Origin 6.1; Northampton, MA, USA).
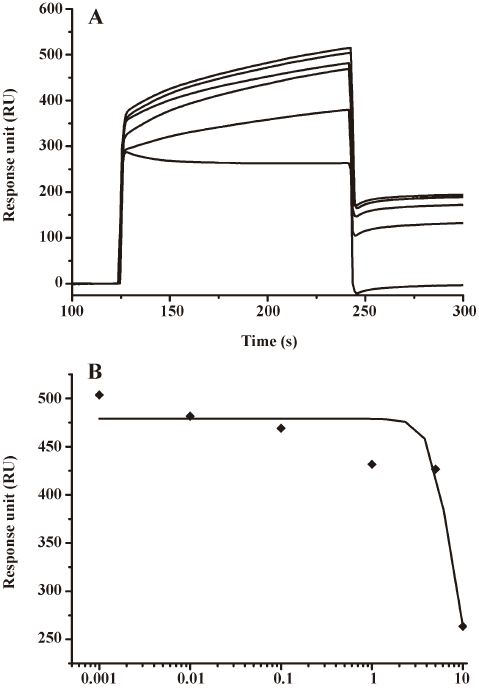
In addition, the SPR binding assay also showed that compound 1 could directly bind to HIV-1 IN at the CCD (Table 1). Therefore, compound 1 inhibited HIV-1 integrase binding to viral DNA by acting as a CCD binder.

Full table
Antiretroviral activity The antiviral activity of compound 1 against the HIV-induced cytopathic effect (CPE) in the C8166 cell culture was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay[20–22]. The results showed that compound 1 could inhibit HIV-1(IIIB) replication by an EC50 value of 17.05 µg/mL in the C8166 cells, and cytotoxicity was observed with a 50% cytotoxic concentration value of 73.11 µg/mL in mock-infected C8166 cells.
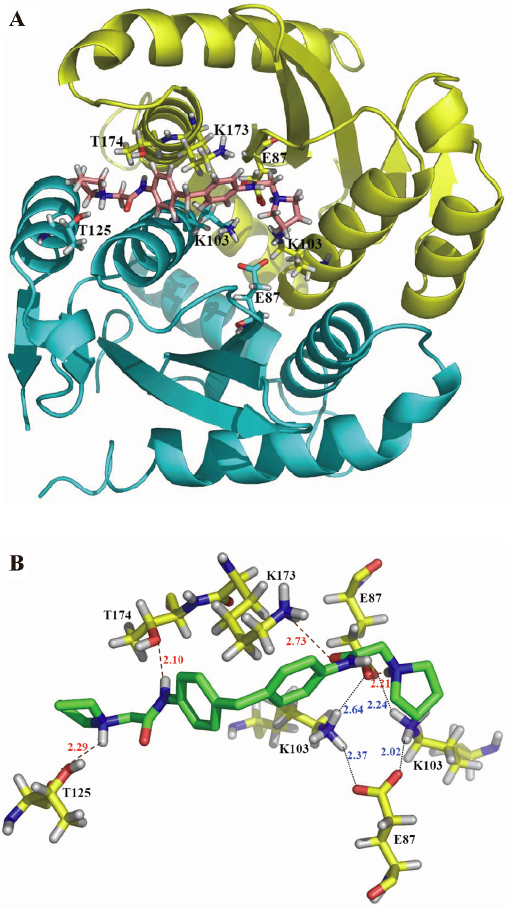
Binding site identified with molecular docking Molecular docking was used to identify the potential binding site of the compound 1 at the HIV-1 IN CCD with PDB code 1QS4. According to the docking results of GOLD (Figure 3A), compound 1 bound to the CCD dimer interface instead of the DDE (D64, D116 and E152) motif. The residues of both chains of the homodimer were involved in the compound 1/IN CCD interaction. As shown in Figure 3B, an N-H bond of compound 1 forms N-H-O hydrogen bond with the O atom of Thr-174 in chain A. This hydrogen bond might greatly determine the orientation of compound 1 in the binding site. The positive-charged side chain of Lys173 in chain A interacts with the right benzene ring of compound 1 by a cation–π interaction. Moreover, a well-defined salt bridge ring exists around the 2 NH3+ groups of Lys103 in both chains. The docking results revealed that residues Thr174, Lys173, and Lys103 in chain A, and Lys-103 in chain B, might play important roles in the interaction between compound 1 and IN CCD.
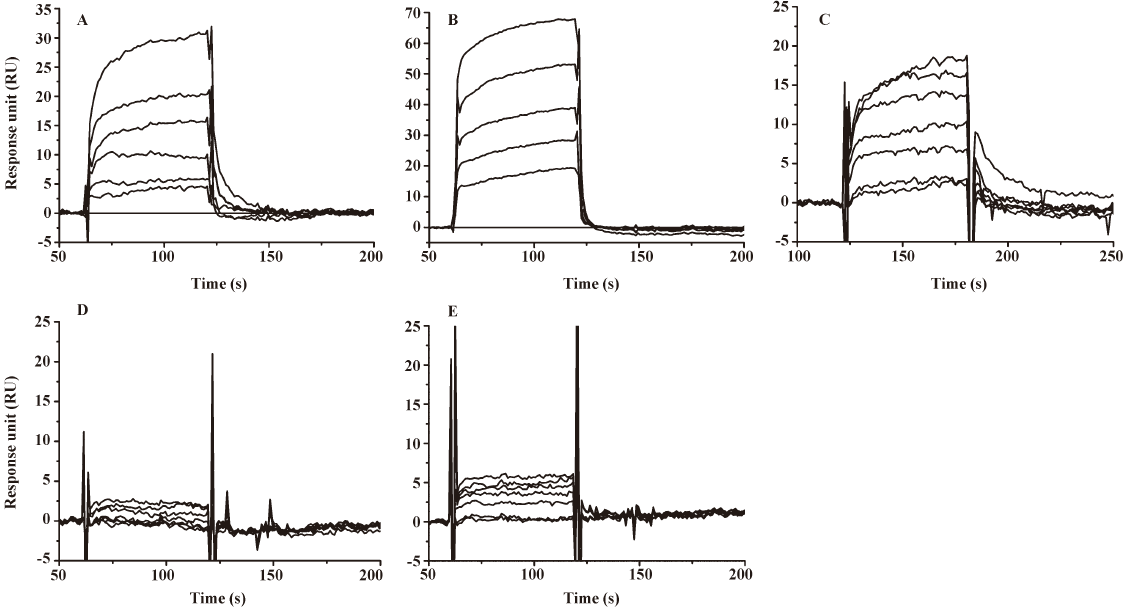
Binding site validated by site-directed mutagenesis To validate the binding site of compound 1 to IN CCD, a site-directed mutagenesis technique-based assay was performed. Three of the important residues which are involved in the interaction between compound 1, and HIV-1 IN CCD were selected for mutation: Lys103 (involved in salt bridge, although it indirectly interacts with compound 1), Lys173 (mainly involved in a cation–π interaction and hydrogen bond), and Thr174 (only involved in hydrogen bond). During the test, Lys103, Lys173, and Thr174 were substituted by alanine, respectively. As indicated in Table 1 and Figure 4, the SPR binding assay suggested that substitution of alanine for Lys103 could significantly reduce the binding of compound 1 to IN, while the alanine mutation for Lys173 and Thr174 (K173A and T174A) almost abolished the binding of compound 1 to IN. Therefore, our mutagenesis experiments confirmed that residues Lys103, Lys173, and Thr174 were involved in the IN interaction with compound 1, supporting the docking results.
Discussion
To date, many IN inhibitors and related binding sites have been reported[14,15,33,34]. However, few have been used in clinical trials so far[35] and many of the IN inhibitors belong to the β-diketo-like acid (DKA) family. Previous research revealed that 2 different binding sites (the donor and the acceptor sites) for DKA may coexist in the active site of IN[34], while in the case of the azidothymidine (AZT) analog, K156, K159, and K160 were identified as key residues involved in nucleotide binding[15]. Moreover, an acetylated-inhibitor binding site K173 was recently reported[33]. It was discovered that the acetylated inhibitor specifically bound at an architecturally-critical region that was located at the IN CCD dimer interface with K173 as a key residue[33].
The current work indicated that compound 1 could inhibit IN binding to viral DNA as a potential IN com-petitive inhibitor. Molecular docking, site-directed mutagenesis and the SPR technology-based assay revealed that compound 1 could bind to the interface of the HIV-1 IN CCD dimer. The ablation of interaction to compound 1 for IN CCD when the residues Lys173 and Thr174 were mutated to alanine illustrates their importance for compound 1/IN CCD interaction. The significant reduction binding affinity between them when the residue Lys103 was mutated to alanine indicates that the salt bridge ring is important for their binding. When considering that CCD contains the catalytic triad and interacts with substrate viral DNA, it could be presumed that compound 1 might prevent DNA binding once bound to IN CCD functioning as an IN inhibitor.
Since it has been identified that 3 key residues, Lys103, Lys173, and Thr174, are involved in the binding of compound 1 to CCD and compound 1 does not contain methyl ester group, we excluded the possibility that it could interact with Lys173 as an acetylated inhibitor as previously reported[33]. Thus it can be tentatively concluded that compound 1 might exhibit a different inhibition mechanism from the above-mentioned cases for acetylated-inhibitor and DKA.
Recently, Li et al reported a peptide IN inhibitor, NL-6, which corresponds to IN residues 97–108[36]. Alanine-scanned analog results showed a decrease in inhibitory potency when Lys103 was substituted by Ala (NL-6-K7A), which indicated that Lys103 might be an important site for IN inhibitor binding[36]. Interestingly, our mutagenesis assay also confirmed that the substitution of Lys103 with Ala could cause an impressive decrease in the binding affinity of compound 1 to IN, thus suggesting that residue Lys103 of IN also plays an important role for compound 1 binding to IN.
In conclusion, we identified a novel compound (compound 1) that could competitively inhibit IN binding to viral DNA. The antivirus assay indicated that compound 1 showed good antiretroviral activity on HIV-induced CPE in MT-4 cell culture. The SPR assay indicated that it bound to the HIV-1 IN CCD domain. Molecular docking provides a possible binding mode for compound 1/HIV-1 IN interaction at the atomic level. Site-directed mutagenesis analysis further identified that 3 key amino acid residues (Lys103, Lys173, and Trp174) were involved in this interaction. Our studies are expected to provide further information in the identification of new drug-binding sites and the elucidation of a potential IN inhibition mechanism, thereby facilitating antiviral agent discovery.
References
- Asante-Appiah E, Skalka AM. HIV-1 integrase: structural organization, conformational changes, and catalysis. Adv Virus Res 1999;52:351-69.
- Cara A, Guarnaccia F, Reitz MS, Gallo RC, Lori F. Self-limiting, cell type-dependent replication of an integrase-defective human immunodeficiency virus type 1 in human primary macrophages but not T lymphocytes. Virology 1995;208:242-8.
- Pommier Y, Neamati N. Inhibitors of human immunodeficiency virus integrase. Adv Virus Res 1999;52:427-58.
- Sherman PA, Fyfe JA. Human immunodeficiency virus integration protein expressed in Escherichia coli possesses selective DNA cleaving activity. Proc Natl Acad Sci USA 1990;87:5119-23.
- LaFemina RL, Callahan PL, Cordingley MG. Substrate specificity of recombinant human immunodeficiency virus integrase protein. J Virol 1991;65:5624-30.
- Bushman FD, Craigie R. Activities of human immunodeficiency virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc Natl Acad Sci USA 1991;88:1339-43.
- Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov 2005;4:236-48.
- Chiu TK, Davies DR. Structure and function of HIV-1 integrase. Curr Top Med Chem 2004;4:965-77.
- Cai M, Zheng R, Caffrey M, Craigie R, Clore GM, Gronenborn AM. Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat Struct Biol 1997;4:567-77.
- Wang JY, Ling H, Yang W, Craigie R. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. Embo J 2001;20:7333-43.
- Dayam R, Deng J, Neamati N. HIV-1 integrase inhibitors: 2003-2004 update. Med Res Rev 2006;26:271-309.
- Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, et al. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000;287:646-50.
- Cotelle P. Patented HIV-1 integrase inhibitors (1998–2005). Recent Pat Anti-infect Drug Disc 2006;1:1-15.
- Johnson AA, Marchand C, Pommier Y. HIV-1 integrase inhibitors: a decade of research and two drugs in clinical trial. Curr Top Med Chem 2004;4:1059-77.
- Drake RR, Neamati N, Hong H, Pilon AA, Sunthankar P, Hume SD, et al. Identification of a nucleotide binding site in HIV-1 integrase. Proc Natl Acad Sci USA 1998;95:4170-5.
- Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, et al. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J Virol 2008;82:764-74.
- Savarino A. In-silico docking of HIV-1 integrase inhibitors reveals a novel drug type acting on an enzyme/DNA reaction intermediate. Retrovirology 2007;4:21.
- Jenkins TM, Engelman A, Ghirlando R, Craigie R. A soluble active mutant of HIV-1 integrase: involvement of both the core and carboxyl-terminal domains in multimerization. J Biol Chem 1996;271:7712-8.
- Du L, Shen L, Yu Z, Chen J, Guo Y, Tang Y, et al. Hyrtiosal, from the marine sponge Hyrtios erectus, inhibits HIV-1 integrase binding to viral DNA by a new inhibitor binding site. Chem Med Chem 2008;3:173-80.
- Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods 1988;20:309-21.
- Johnson VA, Byington RE. Quantitative assays for virus infectivity. New York: Srockton Press; 1990.
- Wang Q, Wang YT, Pu SP, Zheng YT. Zinc coupling potentiates anti-HIV-1 activity of baicalin. Biochem Biophys Res Commun 2004;324:605-10.
- Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity: a rapid access to atomic charges. Tetrahedron 1980;36:3219-28.
- Clark M, Cramer R, van Opdebosch N. Validation of the general purpose Tripos 5.2 force field. J Comput Chem 1989;10:982-1012.
- Sybyl (Computer program). Version 7.0. St Louis, MO: Tripos Associates Inc; 2004.
- Berman HM, Bhat TN, Bourne PE, Feng Z, Gilliland G, Weissig H, et al. The Protein Data Bank and the challenge of structural genomics. Nat Struct Biol 2000;7 Suppl:957-9.
- Goldgur Y, Craigie R, Cohen GH, Fujiwara T, Yoshinaga T, Fujishita T, . Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc Natl Acad Sci USA 1999; 96: 13 040−3.
- Weiner SJ, Kollman PA, Nguyen DT, Case DA. An all atom force field for simulations of proteins and nucleic acid. J Comput Chem 1986;11:431-9.
- Weiner SJ, Kollman PA, Case DA, Singh C, Ghio G, Alagona S, et al. New force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc 1984;106:765-84.
- Rosin CD, Belew RK, Morris GM, Olson AJ, Goodsell DS. Computational coevolution of antiviral drug resistance. Artif Life 1998;4:41-59.
- Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins 2003;52:609-23.
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727-48.
- Shkriabai N, Patil SS, Hess S, Budihas SR, Craigie R, Burke TR Jr, et al. Identification of an inhibitor-binding site to HIV-1 integrase with affinity acetylation and mass spectrometry. Proc Natl Acad Sci USA 2004;101:6894-9.
- Marchand C, Zhang X, Pais GC, Cowansage K, Neamati N, Burke TR Jr, . Structural determinants for HIV-1 integrase inhibition by beta-diketo acids. J Biol Chem 2002; 277: 12 596−603.
- Merck. Monogram and Merck to collaborate on phase III trials of Merck’s investigational HIV-1 integrase inhibitor. [Cited 07 Sep 2007] Available from URL: .http://www.hivandhepatitis.com/recent/2007/090707_a.html
- Li HY, Zawahir Z, Song LD, Long YQ, Neamati N. Sequence-based design and discovery of peptide inhibitors of HIV-1 integrase: insight into the binding mode of the enzyme. J Med Chem 2006;49:4477-86.