
Apoptosis induced by diallyl disulfide in human breast cancer cell line MCF-71
Introduction
Breast cancer is one of the most common malignancies in women and is the leading cause of death worldwide for women between the ages of 40 and 55 years. If effective chemopreventive treatment for breast cancer could be developed, it would have a significant impact on breast cancer morbidity and mortality[1]. However, breast cancer cells are highly resistant to chemotherapy and there is still no effective cure for patients with advanced disease, especially in cases of hormone-independent breast cancer[2,3].
A promising new approach to this problem is the use of garlic. A high intake of garlic is associated with a protective effect against various cancers in humans. Several epidemiological studies suggest that garlic is associated with a decrease of the tumor fatality rate[4]. Garlic oil, or more specifically the diallyl disulfide (DADS) it contains, has recently become more appealing as an anticarcinogenic agent, in part due to its ability to induce apoptosis in vitro[5] and inhibit the formation and growth of tumors in vivo[6,7]. Generally, it is the lipid-soluble organic compounds in garlic that are the most effective antiproliferative agents, and many of them have warranted further study as antitumorigenic agents[8]. Several studies have already demonstrated the efficacy of garlic compounds as antitumorigenic agents. For example, DADS inhibits the in vitro growth of colon, lung and gastric cancer cell lines, and leukemia cell lines[9–11], indicating that DADS may be a useful therapeutic tool in the prevention of cancers. However, a systematic, detailed study of the effects of DADS on human breast cancer cells has yet to be performed. Thus the primary objective of our study was to examine the effect of DADS on human mammary cancer cell line MCF-7.
A secondary objective was to explore the possible mechanisms for any anticancer effects of DADS. One possibility is that DADS promotes apoptosis, and several signaling pathways have been implicated in the regulation of neuronal apoptosis[12]. These include the extracellular signal-regulated protein kinase (ERK) 1/2, the c-Jun NH2-terminal protein kinase (JNK), and the p38 mitogen-activated protein kinase (MAPK). ERK1/2 are preferentially activated by growth factors and neurotrophic factors, while JNK and p38 are preferentially activated by cell stress-inducing signals, such as oxidative stress, environmental stress, and toxic chemical insults[13,14]. Much attention has been focused on the MAPK pathway as a possible target for newly-designed antineoplastic drugs. Therefore, it is useful to establish the role that the MAPK pathway plays in the process that leads to differentiation within a specific tissue or cell type. In our study, ERK, p53, and JNK activity were observed after DADS-induced differentiation in the human breast cancer cell line MCF-7.
Materials and methods
Drugs and reagents The breast cancer cell line MCF-7 was provided by the Cancer Research Institute of Xiangya Medical School of Central South University (Changsha China). DADS was purchased from Sigma (St Louis, MO, USA), RPMI-1640 from Gibco (Grand Island, NY, USA), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) from Amresco (Solon, OH, USA). DMSO, PD98059 (a specific p42/44 MAPK cascade inhibitor), and SDS were purchased from Sigma (USA). Anticaspase-3, JNK, phospho-JNK, ERK, phospho-ERK, phospho-p38, p38, horseradish peroxidase-conjugated second antibody, and enhanced chemiluminescence reagents were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antipoly(ADP-ribose) polymerase (PARP) was obtained from Cell Signaling (Beverly, MA, USA). The annexin V–fluorescein isothiocyanate (FITC) apoptosis detection kit was purchased from Calbiochem (San Diego, CA, USA).
Cell culture The MCF-7 cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum, 1×107 IU·L–1 penicillin, and amphotericin in a 37 oC humidified incubator with an atmosphere of 5% CO2.
Treatments A 50 µmol·L–1 solution of DADS was prepared just before each experiment by dissolving DADS in DMSO. Treatments included different concentrations of DADS ranging from 50 to 400 µmol·L–1 at 37 °C in medium supplemented with serum. For the apoptosis studies, we used 200 µmol·L–1 DADS because that particular concentration induces a significant degree of apoptosis at the times selected, and because it is in the range used for in vivo studies. As a control, an equal amount of DMSO (0.1%) was added to the untreated cells.
Cell viability assay The cells were seeded into 96-well plates and grown to 80% confluence in RPMI-1640. The cultures were then rinsed and incubated with their respective test substances for 24 h. Cell viability was measured using the MTT assay. MTT was dissolved in 1.0 mL phosphate buffered saline (PBS). An amount of this solution equal to 100 mL·L–1 of the culture medium volume was added to the cell cultures. After 6 h of incubation, the cultures were removed from the incubator and the formazan crystals were solubilized by adding DMSO. Metabolic activity was quantified by measuring light absorbance at 570 nm[15].
Ethidium bromide and acridine orange staining The MCF-7 cells treated with different concentrations of DADS were cocultured in 24-well plates for 24 h. PBS (0.9 mL) was mixed with 50 mL ethidium bromide (Sigma, USA) and acridine orange (Amersco, USA) in a 1 mL tube. The cells were then observed under a fluorescence microscope (Olympus, Tokyo, Japan) with a FITC filter set. A total of 200 cells were counted per sample. The percentage of apoptotic cells was calculated by adding the total number of apoptotic living (AL) and apoptotic dead (AD) cells, dividing it by the total number of cells and multiplying it by 100. The percentage of calculated apoptotic cells was as follows:
Apoptosis %=(VA+NVA)/total cell count×100%[16], where VA is viable apoptotic cell, and NVA is non-viable apoptotic cell.
Flow cytometry Cells exposed to different treatments were collected and resuspended in RPMI-1640 medium supplemented with 10% fetal bovine serum. The cell suspension was adjusted to approximately 1×109 cells·L–1. The cells (0.5 mL of the cell suspension) were transferred to a microfuge tube, incubated at room temperature for 15 min in the dark and centrifuged; the media were then removed. After being resuspended in 0.5 mL cold 1×binding buffer [1×104 µmol·L–1 HEPES, 1.5×105 µmol·L–1 NaCl, 2.5×103 µmol·L–1 CaCl2, 1×103 µmol·L–1 MgCl2, and 20% bovine serum albumin (BSA), pH 7.4], 10 µL propidium iodide (4×104 µmol·L–1) was added and the cells were placed on ice and kept away from light. Apoptosis was analyzed by flow cytometry at 488 nm[17].
Western blot analysis After treatment, the cells were removed from the plates and lysed with buffer (5×104 µmol·L–1 Tris–HCl and 5×104 µmol·L–1 NaCl, pH 8.0) and sonicated for 1–2 min on ice. After measuring the protein concentrations, equal amounts of protein from extracts were separated by 15% SDS–PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. Cells with and without treatment were washed with PBS (5×104 µmol·L–1 phosphate, pH 7.4, 1×105 µmol·L–1 NaCl, and 1×104 µmol·L–1 KCl) and lysed on ice for 30 min in PBS containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.5×103 µmol·L–1 phenylmethylsulfonyl fluoride, and 2×104 µmol·L–1 leupeptin. After the lysates were cleared by centrifugation at 15 000×g for 20 min at 4 °C, the total protein was determined using a Bio-Rad protein assay solution (Hercules, CA, USA). Up to 75 g of total protein was denatured in SDS sample loading buffer for 5 min at 95 °C, separated on 10%–15% SDS–PAGE gels, and electro-transferred to Immobilon-P membranes (Millipore, Bedford, MA, USA) using semidry electrophoretic transfer. After the non-specific binding sites on the membranes were blocked with 5% skimmed milk or BSA in TBS-T (2×104 µmol·L–1 Tris-HCl, pH 7.5, 1.37×102 µmol·L–1 NaCl, and 0.01% Tween 20) for 3 h at room temperature with constant shaking, the membranes were incubated in TBST containing the relevant primary antibody (1:500–1000) and 5% BSA overnight at 4 °C. After washing with TBST 3 times, the membranes were incubated in 5% skimmed milk in TBST buffer containing the appropriate second anti-immunoglobulin G antibody (1:5000) at room temperature for 1 h with constant shaking. For the normalization of protein loading, the same membranes were stripped with stripping buffer (1×105 µmol·L–1 2-mercaptoethanol, 2% SDS, and 6.25×104 µmol·L–1 Tris-HCl, pH 6.7) and used for the Western blot assays by the same procedure with a monoclonal antibody against β-actin (1:1000 dilution) and/or with polyclonal antibodies against the relevant protein.
Results
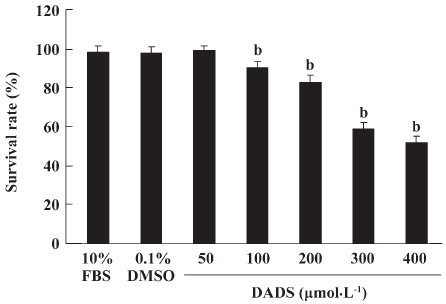
Cell viability The MCF-7 cells were plated at a density of 5000 cells/well into 96-well plates. DADS was added to the wells. The cells were cultured for 24 h and the MTT assay was run. As shown in Figure 1, DADS was found to inhibit the growth of MCF-7 cells in a dose-dependent manner. The treatment of MCF-7 cells for 24 h with 50, 100, 200, 300, or 400 µmol·L–1 DADS resulted in growth inhibition. Survival rates were 99.95%, 91.13%, 83.79%, 59.3%, and 52.56%, respectively (P<0.05; Figure 1).
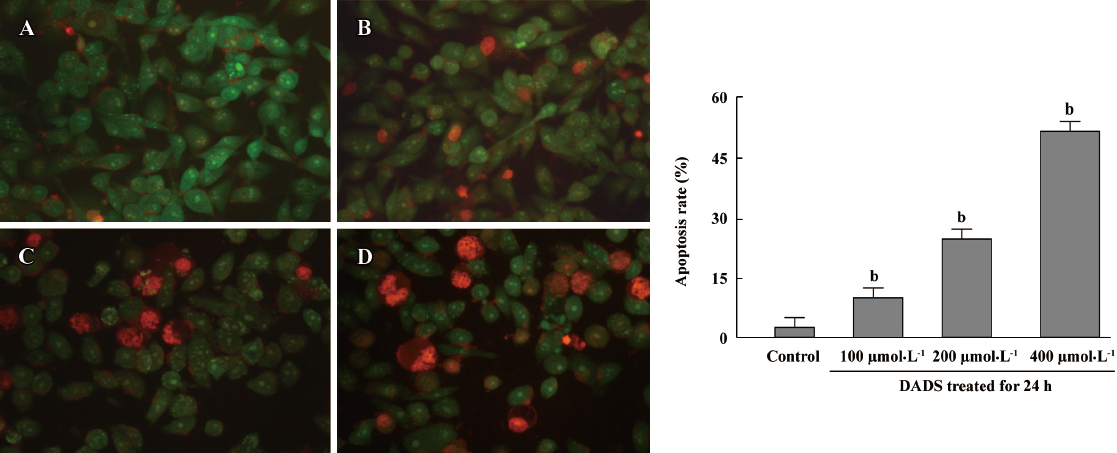
Cell morphology in MCF-7 cells treated by DADS It was previously shown that DADS dose dependently inhibited the growth of MCF-7 cells. To better clarify changes in cell morphology induced by DADS, the MCF-7 cells were treated with DADS at different concentrations of cells and stained with ethidium bromide and acridine orange. Four groups of cells were detected: (1) VNA (green cytoplasm, green unfragmented nucleus); (2) VA (green cytoplasm, green fragmented nucleus); (3) non-viable apoptotic cell (red cytoplasm, red pyknotic nucleus); and (4) non-viable non-apoptotic cell (red cytoplasm, red unfragmented nucleus). The percentage of apoptotic cells was calculated by adding the number of AL and AD cells together, dividing it by the total cell count (200) and multiplying by 100. AL, AD cells were observed. The number of apoptotic MCF-7 cells was significantly increased when the cell suspension was treated for 24 h with 200 µmol·L–1 DADS. The treatment of MCF-7 cells with 100, 200, or 400 µmol·L–1 DADS for 24 h resulted in inducing apoptosis of MCF-7 cells. The apoptosis rates were 9.56%, 18.31%, and 51.23%, respectively (P<0.05 vs control 2.35%; Figure 2).
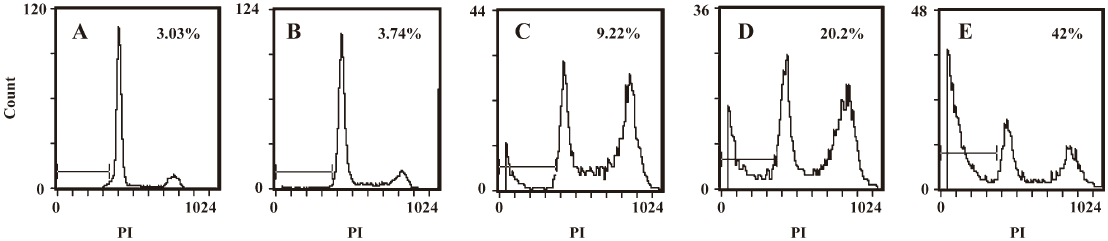
Apoptosis induced by DADS in MCF-7 cells To determine whether DADS induces apoptosis, the MCF-7 cells were treated for 24 h with either 0.1% DMSO or DADS at 50, 100, 200, or 400 µmol·L–1. The cells were collected and stained with propidium iodide and analyzed by flow cytometry. As shown in Figure 3, the proportion of propidium iodide cells increased from 3.74% to 9.22% to 20.2% to 42% in MCF-7 cells treated with the above concentrations of DADS. The value for the control group was 3.03% (Figure 3).
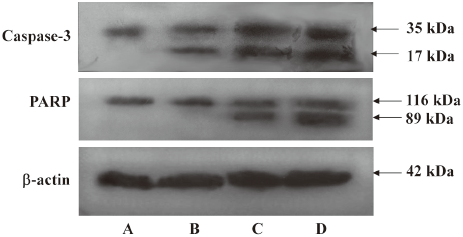
Cleavage of caspase-3 and PARP by DADS in MCF-7 cells In this experiment, the MCF-7 cells were treated with different concentrations of DADS for 24 h. The cells were collected and lysed. Western blot analysis was conducted and probed with antibodies to caspase-3 or to PARP. To further confirm that apoptosis was induced by DADS, caspase-3 and PARP cleavages were detected by Western blot analysis. The results showed that procaspase-3 was cleaved to yield a 17 kDa fragmentation, and PARP was cleaved into an 89 kDa fragment following DADS treatment (Figure 4).
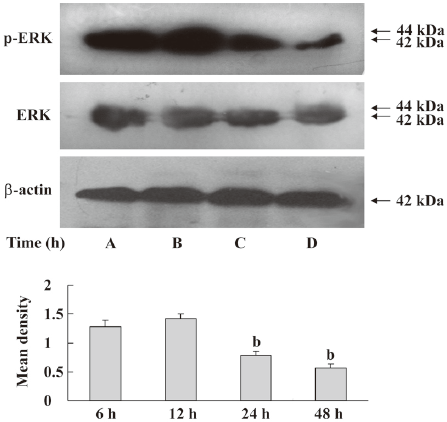
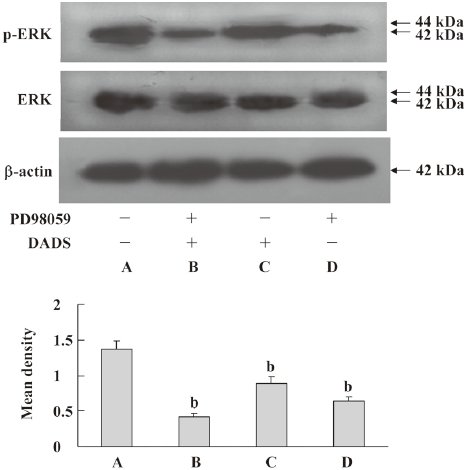
Inhibition of ERK pathway by DADS The MCF-7 cells were treated with 200 µmol·L–1 DADS for 6, 12, 24, or 48 h. The cells were analyzed by Western blotting with antiphospho-ERK or an anti-ERK antibody. We found that phospho-ERK decreased after treatment with DADS. The ERK1/2 protein level did not significantly change in the MCF-7 cells exposed to DADS (Figure 5). PD98059, a specific p42/44 MAPK cascade inhibitor, reinforced the inhibition of phosphorylation by DADS, causing an increase in total ERK1/2 (Figure 6). In parallel experiments, the amount of ERK was determined in the same cell extracts using an antibody that recognized all ERK isoforms, independent of their phosphorylation state.
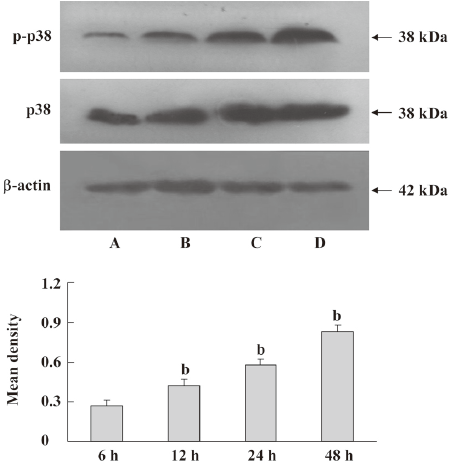
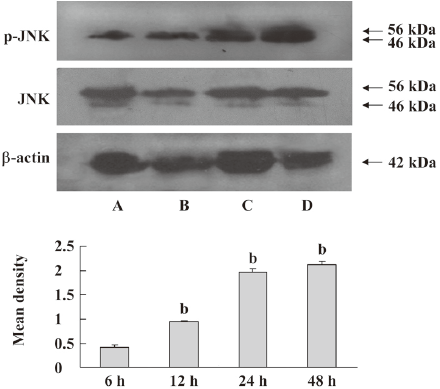
Activation of stress-activated protein kinase/JNK and p38 pathways by DADS The MCF-7 cells were treated with 200 µmol·L–1 DADS for 6, 12, 24, or 48 h. The cells were harvested and lysed. The total protein (40 μg/lane) was loaded on an SDS–PAGE gel, transferred to a PVDF membrane, and probed with antibodies to phospho-p38, p38, phospho-JNK, or JNK. Phospho-p38 and phospho-JNK increased with time. The protein levels of p38 and JNK did not change significantly in the MCF-7 cells exposed to DADS (Figures 7, 8).
Discussion
The objectives of this study were to determine if DADS induced apoptosis in a human breast cancer cell line (MCF-7) and to elucidate the underlying molecular mechanisms. In the present study, DADS was also proven to be an effective inhibitor of human estrogen-receptor ER-positive human breast cells, and our results indicate that DADS dose dependently induces growth inhibition in these cells. The growth inhibitory properties of DADS were also attributed to its induction of apoptotic cell death by the increased proportion of cells in the sub G1 population[18]. The activation of caspase-3 is required for the induction of apoptosis induced by several different stimuli[19,20]. DADS induced the cleavage of capspase-3 and PARP, indicating that caspase-3 was activated during the induction of apoptosis. DADS blocked PARP cleavage and reduced the number of classical apoptotic cell deaths after rotenone treatment. Many of the cells had shrunken nuclei and lacked chromatin fragmentation or condensation into discrete dense clumps[21–24]. Taken together, these results suggest that DADS induces caspase-dependent apoptosis in MCF-7 cells.
Probing possible mechanisms for the above effects of DADS, we considered MAPK and ERK pathways and JNK and p38 signaling. These have a central role in transducing extracellular signals into cellular response[25–27]. ERK is the archetypal pathway of the MAPK superfamily. It is activated in response to a wide variety of growth factors and mitogens, and sustained activation eventually initiates cell proliferation and elicits hallmarks of transformation. Conversely, the inhibition of ERK has been shown to inhibit cell growth and growth factor-stimulated gene transcription. In our system, DADS attenuated the phosphorylation levels of ERK. Thus the inhibition of ERK signaling by DADS might be an attractive explanation for the well-documented biochemical and morphological reverse of many cancer cells into a more benign phenotype following DADS treatment. PD98059 reinforced the inhibition of phosphorylation by DADS causing an increase in total ERK 1/2 levels. This increase may be related to a change in the expression of the ERK gene induced by PD98059. It is now necessary to do a systematic, detailed study of the effect of PD98059 and DADS on human breast cancer cells. DADS inhibits ERK1/2 in human gastric carcinoma MGC803 cells, which may be correlated with protein tyrosine phosphatases, which are frequently upregulated in association with decreased cell proliferation and terminal differentiation.
JNK and p38 signaling are implicated in responses to cellular stress, inflammation, and apoptosis[28–30]. JNK is commonly activated when cells are exposed to various stresses, such as growth factor withdrawal, chemotherapeutics, UV, and TNF-α. Furthermore, it was shown that JNK contributed to apoptotic signal transduction in response to various stressful stimuli. In our apoptotic system, DADS enhanced the phosphorylation levels of JNK and p38 in MCF-7 cells. The prolonged and persistent activation of JNK/stress-activated protein kinase (SAPK) and p38 and the inactivation of ERK induce apoptotic cell death.
In conclusion, these results suggest that DADS inhibits the proliferation of MCF-7 cells and induces apoptosis. The mechanisms may involve the inhibition of ERK and the activation of the SAPK/JNK and p38 pathways.
References
- Baselga J, Mendelsohn J. The epidermal growth factor receptor as a target for therapy in breast carcinoma. Breast Cancer Res Treat 1994;29:127-38.
- Bange J, Zwick E, Ullrich A. Molecular targets for breast cancer therapy and prevention. Nat Med 2001;7:548-52.
- Chopin V, Toillon RA, Jouy N, Bourhis X. P21 (WAF1/CIP1) is dispensable for G1 arrest, but indispensable for apoptosis induced by sodium butyrate in MCF-7 breast cancer cells. Oncogene 2004;23:21-9.
- Agarwal KC. Therapeutic actions of garlic constituents. Med Res Rev 1996;16:111-24.
- Sundaram SG, Milner JA. Diallyl disulfide induces apoptosis of human colon tumor cells. Carcinogenesis 1996;17:669-763.
- Sundaram SG, Milner JA. Diallyl disulfide suppresses the growth of human colon tumor cell xenografts in athymic nude mice. J Nutr 1996;126:1355-61.
- Singh SV, Mohan RR, Agarwal R, Benson PJ, Hu X, Rudy MA, et al. Novel anti-carcinogenic activity of an organosulfide from garlic: inhibition of H-RAS oncogene transformed tumor growth in vivo by diallyl disulfide is associated with inhibition of p21H-ras processing. Biochem Biophys Res Commun 1996;225:660-5.
- Knowles LM, Milner JA. Possible mechanism by which allyl sulfides suppress neoplastic cell proliferation. J Nutr 2001;131:1061-6.
- Druesne N, Pagniez A, Mayeur C. Diallyl disulfide (DADS) increases histone acetylation and p21waf1/cip1 expression in human colon tumor cell lines. Carcinogenesis 2004;25:1227-36.
- Hong YS, Ham YA, Choi JH, Kim J. Effects of allyl sulfur compounds and extract on the expression of Bcl-2, Bax and p53 in non small cell lung cancer cell lines. Exp Mol Med 2000;32:127-34.
- Yuan JP, Wang GH, Ling H, Su Q, Yang YH, Song Y, et al. Diallyl disulfide-induced G2/M arrest of human gastric cancer MGC803 cells involves activation of p38 MAP kinase pathways. World J Gastroenterol 2004;10:2731-4.
- Hetman M, Xia Z. Signaling pathways mediating anti-apoptotic action of neurotrophins. Acta Neurobiol Exp 2000;60:531-45.
- Davis R J. Signal transduction by the JNK group of MAP kinases. Cell 2000;103:239-52.
- Kohno M, Pouyssegur J. Pharmacological inhibitors of the ERK signaling pathway: application as anticancer drugs. Prog Cell Cycle Res 2003;5:219-24.
- Niemann-Jönsson A, Ares MP, Yan ZQ, Bu DX, Fredrikson GN, Brånén L, et al. Increased rate of apoptosis in intimal arterial smooth muscle cells through endogenous activation of TNF receptors. Arterioscler Thromb Vasc Biol 2001;21:1909-14.
- Liu ZZ, Chen JP, Zhao SL, Li CL. Apoptosis-inducing effect of alternol on mouse lymphocyte leukemia cells and its mechanism. Yao Xue Xue Bao 2007;42:1259-65.
- Pang JJ, Xu RK, Xu XB, Cao JM, Ni C, Zhu WL, et al. Hexarelin protects rat cardiomyocytes from angiotensin II-induced apoptosis in vitro. Am J Physiol Heart Circ Physiol 2004;286:1063-9.
- Nakagawa H, Tsuta K, Kiuchi K, Senzaki H, Tanaka K, Hioki K, et al. Growth inhibitory effects of diallyl disulfide on human breast cancer cell lines. Carcinogenesis 2001;22:891-7.
- Zhou ZX, Sun XH, Kang YJ. Ethanol-induced apoptosis in mouse liver Fas- and cytochrome c-mediated caspase-3 activation pathway. Am J Pathol 2001;159:329-38.
- Terutoshi H, Saeko TO, Kaoru T, Yasuo M, Toshio N, Yumi T, et al. Caspase-3 activation by lysosomal enzymes in cytochrome c-independent apoptosis in myelodysplastic syndrome-derived cell line P39. Cancer Res 2001;61:2878-84.
- Kwon KB, Yoo SJ, Ryu DG. Induction of apoptosis by diallyl disulfide through activation of caspase-3 in human leukemia HL-60. Biochem Pharmacol 2002;63:41-7.
- Gunadharini DN, Arunkumar A, Krishnamoorthy G. Antiproliferative effect of diallyl disulfide (DADS) on prostate cancer cell line LNCaP. Cell Biochem Funct 2006;24:407-12.
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001;410:37-40.
- Ling H, Zhang LY, Su Q. Erk is involved in the differentiation induced by diallyl disulfide in the human gastric cancer cell line MGC803. Cell Mol Biol Lett 2006;11:408-23.
- Yuan JP, Wang GH, Ling H. Diallyl disulfide-induced G2/M arrest of human gastric cancer MGC803 cells involves activation of p38 MAP kinase pathways. World J Gastroenterol 2004;10:2731-4.
- Kolbus A, Herr I, Schreiber M. c-Jun-dependent CD95-L expression is a rate-limiting step in the induction of apoptosis by alkylating agents. Mol Cell Biol 2000;20:575-82.
- Hatai T, Matsuzawa A, Inoshita S. Execution of apoptosis signal-regulating kinase I (ASKI)-induced apoptosis by the mitochondria-dependent caspase activation. J Biol Chem 2002; 75: 26 576–81.
- Holloway G, Coulson BS. Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. J Virol 2006; 11: 10 624–33.
- Li XH, Zhang R, Luo DH, Park SJ, Wang Q, Yongsok K, . Tumor necrosis factor α-induced desumoylation and cytoplasmic translocation of homeodomain-interacting protein kinase 1 are critical for apoptosis signal-regulating kinase 1-JNK/p38 activation. J Biol Chem 2005; 15: 15 061–70.
- Hargett D, McLean T, Bachenheimer SL. Herpes simplex virus ICP27 activation of stress kinases JNK and p38. J Virol 2005;13:8348-60.