
Tissue-specific effects of atorvastatin on 3-hydroxy-3-methylglutaryl-coenzyme A reductase expression and activity in spontaneously hypertensive rats1
Introduction
Early experimental evidence[1] revealed that the spontaneously hypertensive rat (SHR) has lower serum cholesterol level than the normotensive Wistar-Kyoto rat (WKY). Since the liver is the major source of circulating cholesterol[2–4], we can assume that cholesterol synthesis in the SHR liver is rather less than that in WKY. However, the exact levels of cholesterol content and 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in cholesterol biosynthesis[5,6], in the liver of SHR remains unknown.
In addition, some reports[7–9] have revealed that the development of hypertension in SHR is accompanied by cardiovascular remodeling, including cardiovascular hypertrophy and enhanced growth of cardiac fibroblasts and vascular smooth muscle cells. Although cholesterol is an integral component of all eukaryotic cells and is essential for normal cellular functions[10,11], its content or local cholesterol synthesis, especially the HMG-CoA reductase expression and activity, may be changed, and this may be associated with the alterations of cell volume and tissue components in cardiac remodeling in SHR.
Several recent studies support the notion that statins attenuate adverse cardiovascular and kidney remodeling in SHR[12–14]. However, little was known about the effect of statins on HMG-CoA reductase itself in the cardiovascular system of SHR. Therefore, we investigated the effects of atorvastatin on tissue cholesterol content and HMG-CoA reductase expression and activity in these tissues.
The purposes of the present study were to characterize the effects of atorvastatin on tissue cholesterol content and HMG-CoA reductase expression and activity in liver, heart, aorta and kidney in SHR.
Materials and methods
Animals and tissue preparation Eight-week-old male SHR and WKY rats were obtained from the Experimental Animal Center, Chinese Academy of Sciences (grade I, Certificate N
Lipid analysis Serum total cholesterol (STC), high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) concentrations were determined by commercial enzymatic methods (test kits from Shanghai Rongsheng Biotech, Inc., Shanghai, China). The cholesterol contents in the tissues (liver, heart, aorta and kidney) were measured after the lipid was extracted as described by Folch[17]. The amounts of cholesterol were determined using the same kit described above.
Western-blot analysis The total proteins were iso-lated from liver, heart, aorta and kidney, and protein concentrations were determined by the Lowry method. An aliquot of 10 µg of protein from each sample was separated on 10% sodium dodecyl sulfate-polyacrylamide gel, electrophoresed, and transferred onto nitrocellulose membranes. The membrane was blocked with Tris-buffered saline (TBS, pH 7.6) containing 5% skim milk and 0.05% Tween-20, and then incubated with anti-HMG-CoA reductase rabbit polyclonal antibodies (1:1000 dilution; Upstate, NY, USA) for 12 h at 4 °C. After the membrane was incubated with goat-anti-rabbit IgG conjugated to horseradish peroxidase (1:5000 dilution; MultiSciences, Hangzhou, China) for 1 h at 37 °C, the immune complexes were visualized by the enhanced chemiluminescence (ECL) method. Quantification of the bands was carried out using densitometric analysis software Quantity One (Bio-Rad, CA, USA). To ensure equal protein loading, GAPDH was used as an endogenous control.
HMG-CoA reductase activity determination Tissue (liver, heart, aorta or kidney) endoplasmic reticulum (ER) preparation, HMG-CoA reductase assay, and one-step isolation of mevalonolactone were carried out as previously described[18]. Briefly, the calcium precipitation technique was used to prepare endoplasmic reticulum, and the microsomal protein concentrations were determined by the Lowry method. Incubations for enzyme assay were carried out in a total volume of 150 µL, which contained 0.1–0.8 mg of microsomal protein, 2 mmol/L, NADPH, 30 mmol/L glucose-6-phosphate, 1.75 IU/mL glucose-6-phosphate dehydrogenase, and 0.2 mmol/L HMG-CoA (all from Sigma, St Louis, MO, USA). The reaction time was 45 min, and the samples were incubated for another 30 min at 37 °C to allow complete lactonization of mevalonate. Then mevalonolactone was isolated from the incubation mixture by two successive extractions with 7.5 mL benzene. A 10 mL aliquot of the pooled benzene extracts was trans-ferred to a cryovial and dried at –56 °C in a freeze-drier (Christ Alpha 1-2, PA, USA). Dried mevalonolactone was dissolved in high performance liquid chromatogram (HPLC)-grade water, and the concentration was determined using HPLC[19] (Agilent 1100 HPLC System; Agilent, CA, USA). Chromatographic separations were carried out using an Agilent ZORBA Extend-C18 column (4.6×1.50 cm inner diameter, 3.5 µm; Agilent, CA, USA). Aliquots (100 µL) of each sample were injected into the HPLC system. The mobile phase consisted of HPLC-grade water, and the elution was carried out at 1 mL/min at 37 °C. The detection was carried out at 200 nm. HMG-CoA reductase activity was determined by calculating the concentrations of mevalonolactone and expressed as units per gram of protein (1 unit yields 1 µmol of product per min).
Statistical analysis Results are represented as mean±SD. Statistical analysis was carried out with SPSS 13.0 statistical software. One-way ANOVA followed by Bonferroni post hoc test was used to determine significant differences between multiple groups. The significance level was set at P<0.05.
Results
Serum lipid content The SHR control group showed lower STC and LDL-C levels (P<0.05 for each, Table 1) but a similar value of HDL-C compared with the WKY-control group. Long-term treatment with atorvastatin led to a clear reduction of STC and LDL-C in both strains (Table 1).
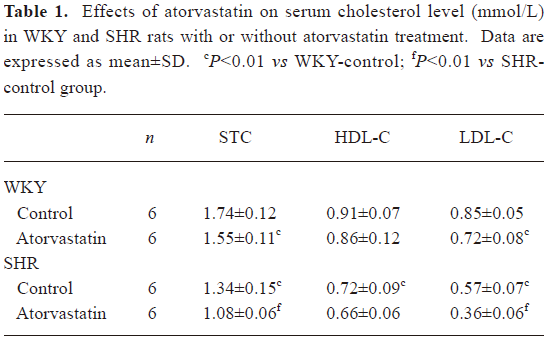
Full table
Tissue cholesterol profile In liver, cholesterol concentrations were remarkably lower in the SHR-control than in the WKY-control group (Table 2, P<0.01). However, in the other three tissues (heart, aorta and kidney), cholesterol levels were comparable between SHR and WKY control groups. Treatment with atorvastatin influenced cholesterol content only in SHR. In the SHR-atorvastatin group, drug intervention significantly reduced the cholesterol level in liver (P<0.01), heart (P<0.05), aorta (P<0.05), and kidney (P<0.05) compared with the SHR-control group (Table 2).
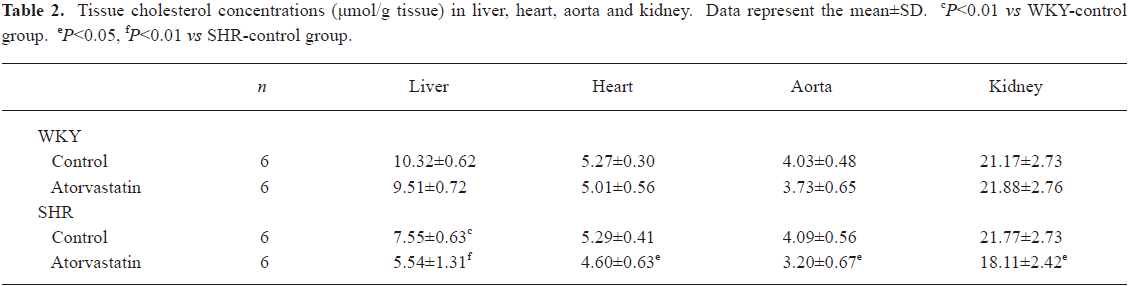
Full table
Effect of atorvastatin on HMG-CoA reductase expression and activity The baselines of HMG-CoA reductase expression and activity in all four tissues were strikingly higher in the SHR-control than in the WKY-control group (Figure 1). In WKY, atorvastatin treatment changed HMG-CoA reductase only in the liver, where it remarkably increased the protein expression (P<0.01 vs WKY-control) but reduced the enzyme activity (P<0.05 vs WKY-control). These indices were not changed in the other three extrahepatic tissues from the WKY-atorvastatin group. However, both the protein expression and the enzyme activity decreased after 10 weeks of atorvastatin administration in these four tissues of SHR rats.
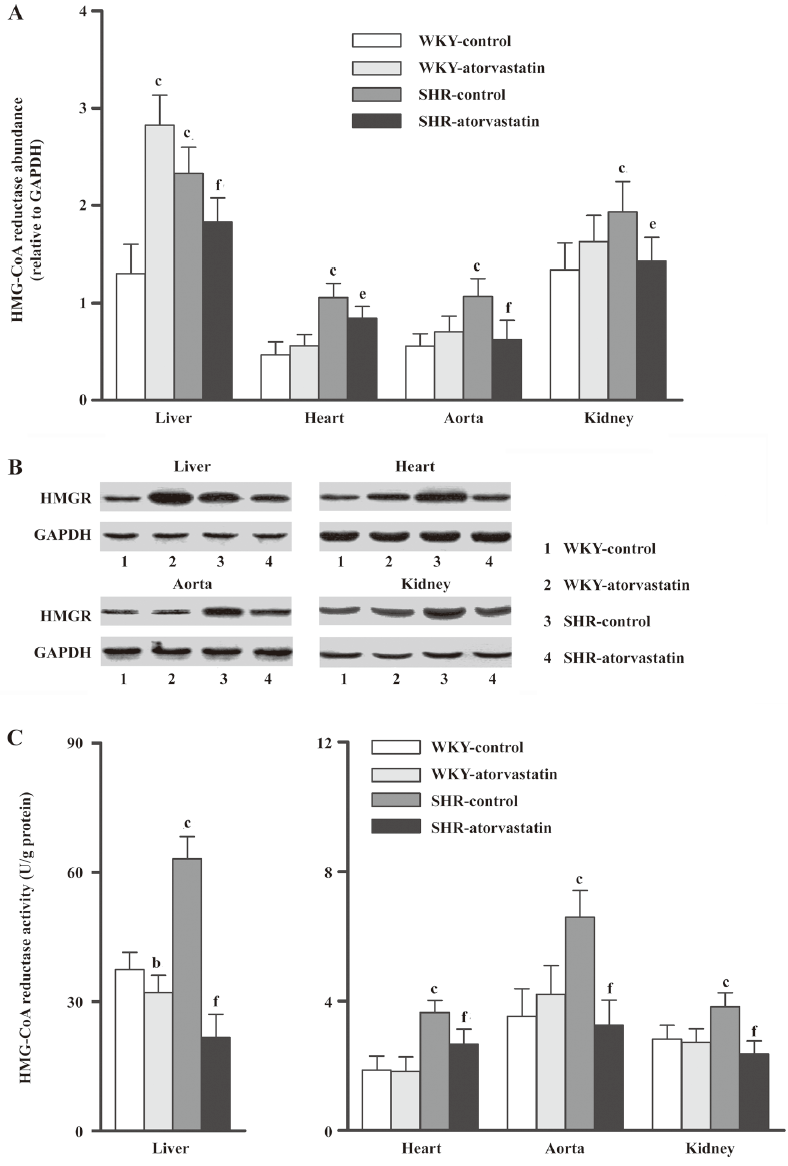
Discussion
Liver cholesterol is mainly derived from serum LDL, which enters hepatocytes by receptor-mediated endocytosis, as well as from local synthesis within the liver[20] and absorption of dietary cholesterol. Meanwhile, HMG-CoA reductase is the rate-limiting enzyme in local cholesterol biosynthesis[5,6], and the liver is the major source of circulating cholesterol[2–4]. In addition, HMG-CoA reductase expression and activity is modulated through negative feedback regulation by the end product, cholesterol[20].
First, we found that the SHR-control group had lower STC and liver cholesterol contents than the WKY-control group, which was consistent with previous reports that the synthesis of liver cholesterol was abnormally decreased in SHR[1]. Moreover, previous reports[21,22] revealed that the lower activity of mevalonate pyrophosphate decarboxylase, an important enzyme locating downstream of HMG-CoA reductase, was responsible for the reduced cholesterol synthesis in SHR liver. As a result of lower cholesterol synthesis within the SHR liver, hepatic cholesterol content and serum cholesterol were lower, and correspondingly the HMG-CoA reductase expression and activity were upregulated, probably through negative feedback by the end product, cholesterol[20].
Second, our study revealed that atorvastatin, a competitive HMG-CoA reductase inhibitor, caused a large compensatory induction of hepatic HMG-CoA reductase, but a significant depression of enzyme activity in the WKY liver. This result might be related to the fact that atorvastatin or its active metabolites resides longer in hepatocytes than other statins[23,24]. Therefore, the HMG-CoA reductase activity was competitively inhibited not only in vivo, but also ex vivo when it was determined in our experiment. Thus, the total enzyme activity obtained by the ex vivo test was depressed despite the greater amount of protein. In WKY liver, atorvastatin not only competitively inhibits HMG-CoA reductase activity and cholesterol synthesis[25], but also increases the number of hepatic LDL receptors to enhance the uptake and catabolism of circulating LDL[26–28]. Mainly through these two mechanisms, circulating cholesterol decreased in the WKY-atorvastatin group. Subsequently, HMG-CoA reductase expression was enhanced, also mediated through end-product feedback[20]. On the other hand, because of the compensatory upregulation of HMG-CoA reductase and the enhanced hepatic uptake of LDL, the content of liver cholesterol in the WKY-atorvastatin group did not drop sharply. In addition, several reports[29,30] revealed that statins increased intestinal cholesterol absorption in humans and animals. This is probably one of the reasons why cholesterol content in the liver of WKY was not affected by atorvastatin.
Third, both the activity and the expression of HMG-CoA reductase were reduced by atorvastatin treatment in SHR liver. The level of HMG-CoA reductase is controlled by many interactions, including synthesis and degradation of the protein, which can result in changes of over 200-fold in intracellular levels of the enzyme[20]. Therefore, we suggest following mechanisms: (1) SHR might have defects in end product feedback mechanism of hepatic HMG-CoA reductase; and (2) SHR might have an enzyme conformation more vulnerable to atorvastatin, with a faster degradation rate, since several studies[25,31–32] have shown that statins modify the conformation of HMG-CoA reductase and hence its degradation. Therefore, we can infer that in the livers of the SHR-atorvastatin group, degradation of HMG-CoA reductase may exceed the enzyme compensatory synthesis induced by atorvastatin, and the amount of HMG-CoA reductase decreased. Due to the decrease of HMG-CoA reductase and the prolonged residence of atorvastatin in the hepatocytes[23,24], enzyme activity declined even more, and the liver cholesterol content dropped markedly.
Fourth, data from our study revealed that the SHR-control group had lower serum LDL levels and comparable cholesterol content of extrahepatic tissues (heart, aorta and kidney). We also found that the HMG-CoA reductase expression and activity of these tissues were upregulated. Extrahepatic tissues also have local cholesterol synthesis[33], but under physiological conditions, serum LDL probably supplies their cholesterol needs[34,35]. These results suggest that the supply of LDL to extrahepatic tissues may be insufficient due to low circulating LDL, and subsequently, local cholesterol synthesis increased, probably via upregulation of HMG-CoA reductase expression and activity.
Finally, HMG-CoA reductase expression and activity did not change in extrahepatic WKY tissues after ator-vastatin treatment. These tissues may take up enough LDL from the circulation, despite the decreased serum cholesterol level after atorvastatin treatment. Since the HMG-CoA reductase in these tissues was relatively inactive, atorvastatin had little effect on the enzyme. However, we found that HMG-CoA reductase expression and activity of extrahepatic SHR tissues decreased after atorvastatin treatment. This was consistent with the results from the livers of rats in the SHR-atorvastatin group, indicating the inhibition of local cholesterol synthesis after atorvastatin treatment. Because the local cholesterol synthesis was depressed by atorvastatin, and the circulating LDL supply was insufficient, the cholesterol content in extrahepatic tissues of the SHR-atorvastatin group declined.
In conclusion, this study demonstrated that: (1) both the liver and the extrahepatic tissues of SHR showed abnormal cholesterol biosynthesis where the key enzyme, HMG-CoA reductase, was in an abnormally upregulated state; and (2) the effects of atorvastatin on tissue cholesterol content and HMG-CoA reductase are strain- and tissue-specific.
Limitations Although this study proposed several possible reasons why the effects of statin on HMG-CoA reductase were too different between SHR and WKY, the exact mechanisms should be explored in further study. Also, to understand more precisely the effects of atorvastatin on tissue cholesterol content, morphological examination of these tissues are needed to provide more solutions. In addition, the intestinal source of cholesterol was not investigated in our study and further study will be necessary to further clarify the relevance of the findings of the study.
Acknowledgements
We express our sincere gratitude to Prof Iain C BRUCE (University of Hong Kong) for checking the English. We also thank Jian-zhong SHENTU from the Department of Clinical Pharmacology, The First Affiliated Hospital, College of Medicine, Zhejiang University for his excellent technical support on HPLC.
References
- Iritani N, Fukuda E, Nara Y, Yamori Y. Lipid metabolism in spontaneously hypertensive rats (SHR). Atherosclerosis 1977;28:217-22.
- Gould RG. Lipid metabolism and atherosclerosis. Am J Med 1951;11:209-27.
- Friedman M, Byers SO, Michaelis F. Production and excretion of cholesterol in mammals. IV. Role of liver in restoration of plasma cholesterol after experimentally induced hypocholesteremia. Am J Physiol 1951;164:789-91.
- Hotta S, Chaikoff IL. The role of the liver in the turnover of plasma cholesterol. Arch Biochem 1955;56:28-37.
- Dietschy JM, Brown MS. Effect of alterations of the specific activity of the intracellular acetyl CoA pool on apparent rates of hepatic cholesterogenesis. J Lipid Res 1974;15:508-16.
- Kennelly PJ, Rodwell VW. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase by reversible phosphorylation-dephosphorylation. J Lipid Res 1985;26:903-14.
- Doggrell SA, Brown L. Rat models of hypertension, cardiac hypertrophy and failure. Cardiovasc Res 1998;39:89-105.
- Thorin-Trescases N, Deblois D, Hamet P. Evidence of an altered in vivo vascular cell turnover in spontaneously hypertensive rats and its modulation by long-term antihypertensive treatment. J Cardiovasc Pharmacol 2001;38:764-74.
- Bell D, Kelso EJ, Argent CC, Lee GR, McDermott BJ. Temporal characteristics of cardiomyocyte hypertrophy in the spontaneously hypertensive rat. Cardiovasc Pathol 2004;13:71-8.
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997;89:331-40.
- Howland DS, Trusko SP, Savage MJ, Reaume AG, Lang DM, Hirsch JD, . Modulation of secreted beta-amyloid precursor protein and amyloid beta-peptide in brain by cholesterol. J Biol Chem 1998; 273: 16 576–82.
- Bezerra DG, Mandarim-de-Lacerda CA. Beneficial effect of simvastatin and pravastatin treatment on adverse cardiac remodeling and glomeruli loss in spontaneously hypertensive rats. Clin Sci (Lond) 2005;108:349-55.
- Lee TM, Lin MS, Chou TF, Tsai CH, Chang NC. Effect of pravastatin on development of left ventricular hypertrophy in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 2005;289:H220-7.
- Yang L, Gao YJ, Lee RM. The effects of quinapril and atorvastatin on artery structure and function in adult spontaneously hypertensive rats. Eur J Pharmacol 2005;518:145-51.
- Cilla DD, Whitfield LR, Gibson DM, Sedman AJ, Posvar EL. Multiple-dose pharmacokinetics, pharmacodynamics, and safety of atorvastatin, an inhibitor of HMG-CoA reductase, in healthy subjects. Clin Pharmacol Ther 1996;60:687-95.
- Dostal LA, Whitfield LR, Anderson JA. Fertility and general reproduction studies in rats with the HMG-CoA reductase inhibitor, atorvastatin. Fundam Appl Toxicol 1996;32:285-92.
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem 1957;226:497-509.
- Goodwin CD, Margolis S. Improved methods for the study of hepatic HMG-CoA reductase: one-step isolation of mevalonolactone and rapid preparation of endoplasmic reticulum. J Lipid Res 1976;17:97-103.
- Buffalini M, Pierleoni R, Guidi C, Ceccaroli P, Saltarelli R, Vallorani L, et al. Novel and simple high-performance liquid chromatographic method for determination of 3-hydroxy-3-methylglutaryl-coenzyme A reductase activity. J Chromatogr B Analyt Technol Biomed Life Sci 2005;819:307-13.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990;343:425-30.
- Sawamura M, Nara Y, Yamori Y. Liver mevalonate 5-pyrophosphate decarboxylase is responsible for reduced serum cholesterol in stroke-prone spontaneously hypertensive rat. J Biol Chem 1992;267:6051-5.
- Michihara A, Sawamura M, Nara Y, Ikeda K, Yamori Y. Lower mevalonate pyrophosphate decarboxylase activity is caused by the reduced amount of enzyme in stroke-prone spontaneously hypertensive rat. J Biochem 1998;124:40-4.
- Naoumova RP, Dunn S, Rallidis L, Abu-Muhana O, Neuwirth C, Rendell NB, et al. Prolonged inhibition of cholesterol synthesis explains the efficacy of atorvastatin. J Lipid Res 1997;38:1496-500.
- Ness GC, Chambers CM, Lopez D. Atorvastatin action involves diminished recovery of hepatic HMG-CoA reductase activity. J Lipid Res 1998;39:75-84.
- Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001;292:1160-4.
- Brown MS, Goldstein JL. Lipoprotein receptors in the liver. Control signals for plasma cholesterol traffic. J Clin Invest 1983;72:743-7.
- Goldstein JL, Brown MS. Lipoprotein receptors and the control of plasma LDL cholesterol levels. Eur Heart J 1992; 13 Suppl B: 34–6.
- Kesaniemi YA, Witztum JL, Steinbrecher UP. Receptor-mediated catabolism of low density lipoprotein in man. Quantitation using glucosylated low density lipoprotein. J Clin Invest 1983;71:50-9.
- Miettinen TA, Gylling H. Synthesis and absorption markers of cholesterol in serum and lipoproteins during a large dose of statin treatment. Eur J Clin Invest 2003;33:976-82.
- Briand F, Serisier S, Krempf M, Siliart B, Magot T, Ouguerram K, et al. Atorvastatin increases intestinal cholesterol absorption in dogs. J Nutr 2006;136:2034S-6S.
- Cappel RE, Gilbert HF. The effects of mevinolin on the thiol/disulfide exchange between 3-hydroxy-3-methylglutaryl-coenzyme A reductase and glutathione. J Biol Chem 1989;264:9180-7.
- Istvan ES, Palnitkar M, Buchanan SK, Deisenhofer J. Crystal structure of catalytic portion of human HMG-CoA reductase: insights into regulation of activity and catalysis. EMBO J 2000;19:819-30.
- Jurevics HA, Morell P. Sources of cholesterol for kidney and nerve during development. J Lipid Res 1994;35:112-20.
- Landon EJ, Greenberg DM. Endogenous cholesterol metabolism in the rat studied with C14-labeled acetate. J Biol Chem 1954;209:493-502.
- Turley SD, Andersen JM, Dietschy JM. Rates of sterol synthesis and uptake in the major organs of the rat in vivo. J Lipid Res 1981;22:551-69.