
Development of efficient RNA interference system using EGF-displaying phagemid particles
Introduction
In previous studies, scientists have successfully modified the filamentous bacteriophages, which are capable of delivering genes to mammalian cells using targeting ligands, such as growth factors[1–5], antibodies[6], and viral capsid proteins[7]. To improve the ligand display level on phages, we developed LMP cells (Escherichia coli bearing the ligand-pIII-encoding helper phage) to make cell-targeted phagemid particles[8]. We also demonstrated that it can deliver reporter genes into target cells.
RNAi techniques are a promising way to silence therapeutic target genes. Both non-viral vectors and viral vectors have been used as a vehicle to deliver siRNA in vitro and in vivo. The major problem of non-viral vectors lies in its low gene delivery efficiency and hard quality control. Animal viruses, such as adenoviruses, have broad natural tropism which makes them difficult to selectively transfer siRNA genes into target cells. In contrast to these animal viruses, filamentous bacteriophage has no natural tropism to mammalian cells. However, it can highly efficiently infect the targeted cells when the peptide ligand was displayed on the surface. Like non-viral vectors, the major problem of this vector is its low gene delivery efficiency. For-tunately, hydroxycamptothecin (HCPT), a topoisomerase I inhibitor and also anticancer drug, can substantially improve its gene delivery efficiency[1,13,17,18]. In this study, we tested whether cell-targeted phages can be used as siRNA gene delivery tools or not.
Materials and methods
Reagents Restriction endonucleases were purchased from TaKaRa Biotechnology (Dalian, China). Dulbecco’s modified Eagle’s medium (DMEM), Dulbecco’s phosphate-buffered saline, fetal bovine serum (FBS), and trypsin/ethylenediamine tetraacetic acid were purchased from Invitrogen (Grand Island, NY, USA).
Cell culture H1299 (large-cell lung carcinoma; ATCC, Manassas, VA, USA) cells were cultured at 37 ºC in medium consisting of 90% DMEM and 10% FBS in a humidified atmosphere of 95% air and 5% CO2.
Plasmid construction All oligonucleotides were synthesized by Sangon (Shanghai, China) and annealed to be double-stranded, as previously described[10]. The selection of the coding sequences of EGFP and Akt for siRNA was based on 2 different publications, respectively[9,10]. Detailed information of the oligonucleotides is shown in Table 1.
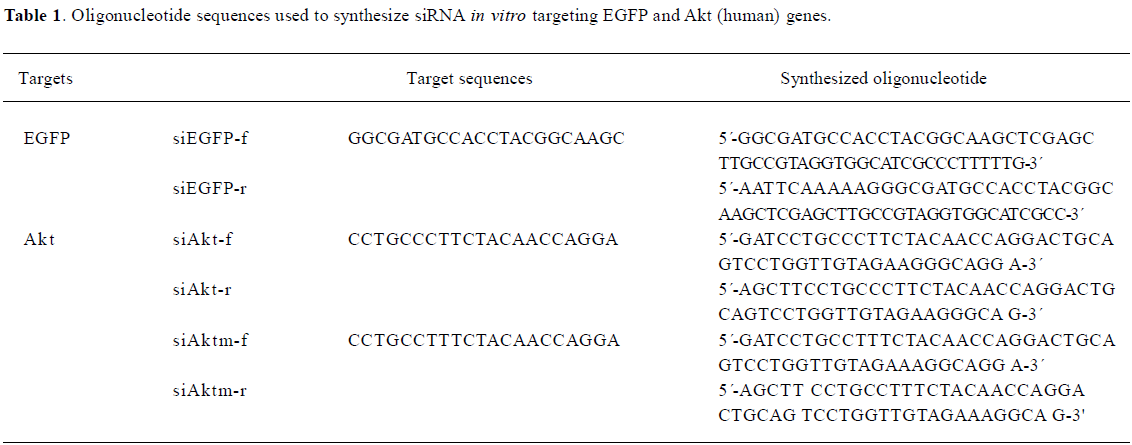
Full table
To generate the pSi1.0-EGFP RNAi plasmid, a 55-nt oligo-DNA duplex (annealed by siEGFP-f and siEGFP-r) corresponding to nucleotides 106–127 of the pEGFP-N1 (BD Biosciences Clontech, Palo Alto, CA, USA)-coding region was directly inserted into the pSilencer1.0 (Ambion, Austin, TX, USA) vector digested with ApaI (blunted) and HindIII.
The pSilencer4.0-f plasmid was generated as follows: the pGl3-control plasmid (Promega category no.: E1741; Promega, Madison, WI,USA) was digested with ScaI, and the 1.08 kb fragment were used to ligate with pSilencer4.0 (Ambion, Austin, TX, USA) which is linearized with ScaI. The inserted subclones with the reversed orientation of cytomegalovirus (CMV) promoter are genetically selected with the supplementary of ampicillin resistance. This modified psilencer4.0 was prepared for the phage package and siRNA expression.
To generate the pSi4.0-Akt plasmid, a 52 nt oligo-DNA duplex (annealed by siAkt-f and siAkt-r) corresponding to nucleotides 1462–1482 of the Akt1 (NM_001014432) and 1244–1264 of Akt2 (NM_001626)-coding region was directly inserted into the pSilencer4.0 vector digested with BamHI and HindIII. The mock Akt sequence is only one nucleotide different from siAkt, as listed in Table 1.
Preparation of phagemid particles The phagemid particles were prepared following previously published protocols[11] with some modifications. M13KO7EGFCT[8] was transformed into Escherichia coli to make LMP cells. The phagemid carrying the siRNA-encoding sequence was transformed into LMP cells. The cells were then plated on LB agar (containing 70 μg/mL kanamycin and 50 μg/mL ampicillin) and incubated at 37 ºC overnight, A cell clone was picked the next day and transferred into 1 L LB containing 70 μg/mL kanamycin and 50 μg/mL ampicillin. After shaking at 37 ºC for 14–16 h, the supernatant of the culture was collected and the phagemid particles were purified with polyethylene glycol (PEG)/NaCl precipitation.
Quantification of phagemid particles The phagemid particles were quantified by ELISA, as previously described[12]. Briefly, serial dilutions of phagemid particles in coating buffer (0.1 mol/L NaHCO3, pH 9.1) were coated in microtiter plates (Corning; Corning, NY, USA) at 4 ºC overnight. After blocking with 1% bovine serum albumin (BSA; Sigma, St Louis, MO, USA) in phosphate-buffered saline (PBS), the bound phages were stained with a mouse anti-M13 antibody conjugated with horseradish peroxidase (HRP; Amersham Biosciences, Piscataway, NJ, USA) diluted at 1:200 in PBST (PBS containing 0.1% Tween-20). The signal was developed using the 2,2´-azino-di-(3-ethylbenzthiazoline-6-sulfonic acid) substrate and quantitated in an ELISA reader (Bio-Rad Laboratories, Hercules, CA, USA). The M13KO7 helper phage of a known number of pfus was used for standardization.
Purification of single-strand DNA from phagemid particles Single-strand DNA (ssDNA) was extracted from the phagemid particles following the supplied protocol (Ph.D.-12 phage display peptide library kit, New England Biolabs, Beverly, MA, USA). Briefly, the phagemid particles were precipitated by PEG/NaCl. The pellet was then suspended with 100 μL iodide buffer. In total, 250 mL ethanol was added and incubated for 10 min to allow for ssDNA precipitation. The pellet was collected after centrifugation at 12 000×g for 10 min, washed in 70% ethanol, and air dried for 10 min. It was finally dissolved in 30 μL TE buffer (10 mmol/L Tris-HCl [pH 8.0] and 1 mmol/L EDTA) and analyzed by agarose gel electrophoresis.
In vitro phagemid particle transfection The cells were plated into 24-well plates at densities of 10 000 cells per well, 24 h prior to the phage particle addition. The phages were added at 1011 pfu/mL and incubated with the cells for 48 h at 37 ºC in complete media. The cells were visualized under an epifluorescent inverted microscope (IX71; Olympus, Tokyo, Japan). All the transfections were done in triplicate and performed at least twice.
HCPT treatment HCPT treatments were performed 48 h after the addition of the phages in medium containing 10% FBS. The medium was removed at 48 h after the phage addition, and the cells were incubated with HCPT at 2.5 μmol/L for 6 h at 37 ºC. This was followed by replacement with fresh medium and an additional incubation of 18 h at 37 ºC.
Western blot analysis In total, 106 H1299 cells transfected transiently with a variety of phagemid particles were lysed after HCPT treatment, as described in the Results. Prior to the lyses, the cells were washed with PBS and collected by scraping. Then, they were lysed in ice-cold Tris buffer (50 mmol/L, pH 7.5) containing 5 mmol/L EDTA, 300 mmol/L NaCl, 0.1% Igepal, 0.5 mmol/L NaF, 0.5 mmol/L Na3VO4, 0.5 mmol/L phenylmethylsulfonyl fluoride, and antiprotease mixture (Roche Molecular Biochemicals, Indianapolis, IN, USA), sonicated, and centrifuged at 13 000×g for 10 min. The supernatant was used for protein determination by the Bradford procedure (Bio-Rad, USA) and Western blotting. The proteins were resolved on 12% SDS-PAGE, transferred onto nitrocellulose membranes, and incubated with the appropriate antibodies. The following protocols are the same as described earlier. The anti-pSer473-AKT antibody (Cell Signaling Technology, Beverly, MA, USA) was used in 1:1000 dilutions. The goat antimouse immunoglobulin G conjugated with HRP (ImmuClub Labs, Sunnyvale, CA, USA) was diluted at 1:10 000 in 1% BSA in PBST. After 1 h incubation, the filters were washed with PBST, developed using a SuperSignal West Pico kit (Pierce Biotechnologies, Rockford, IL, USA) for 5 min, and exposed to X-ray film.
Results
Construction of siRNA expression vectors To improve the expression of siRNA against Akt, the pSilencer4.1-CMV vector which carries the CMV promoter was used. pSi4.1CMV-f1 was constructed by inserting the F1 origin sequence into pSilencer4.1-CMV. pSilencer4.0-siAKT was obtained by insert shRNA against Akt into pSi4.1CMV-f1.
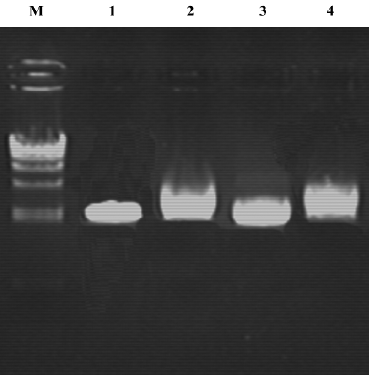
ssDNA analysis To examine the ratio of phagemids to helper phage genomes packaged in the phagemid particles, we analyzed the ssDNA. The results indicate that almost all of the DNA packaged were phagemids (Figure 1).
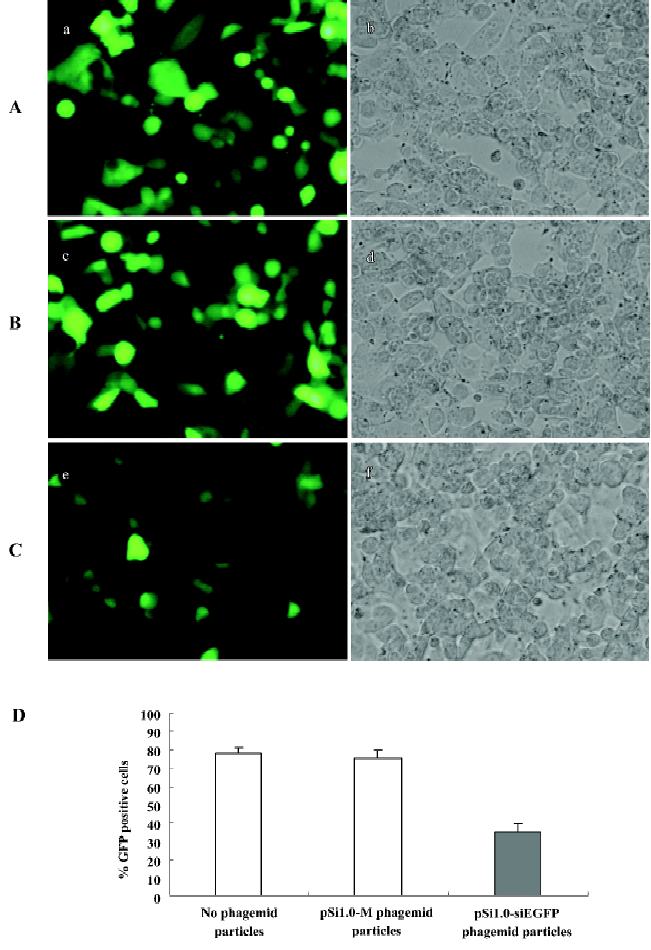
In vitro inhibition of EGFP expression by EGF displaying pSil.0-siEGFP phagemid particle infection To test whether phagemid particles carrying siRNA against EGFP can inhibit EGFP expression, we transfected the pEGFP-N1 plasmid into NCI-H1299 cells. At 12 h after transfection, the phagemid particles (1011 pfu/mL) were transduced into the cells. At 24 h after transduction, HCPT was added into the culture at a final concentration of 2.5 μmol/L; the cells was incubated for another 24 h. The data shown in Figure 2 indicate that pSil.0-siEGFP can significantly inhibit the expression of EGFP.
Phagemid particles of pSi4.1-siAkt can inhibit Akt expression of NCI-H1299 in the presence of HCPT The NCI-H1299 cells was infected with pSi4.1-siAkt phagemid particles packaged by M13KO7EGFCT. HCPT was added to improve the siRNA expression. Western blotting was performed to examine the Akt expression.
As shown in Figure 3A, the pSi4.1-siAKT plasmid transfected by Lipofectamine can inhibit the Akt expression by approximately 50%. No inhibition was found in mock vector pSi4.1-M.
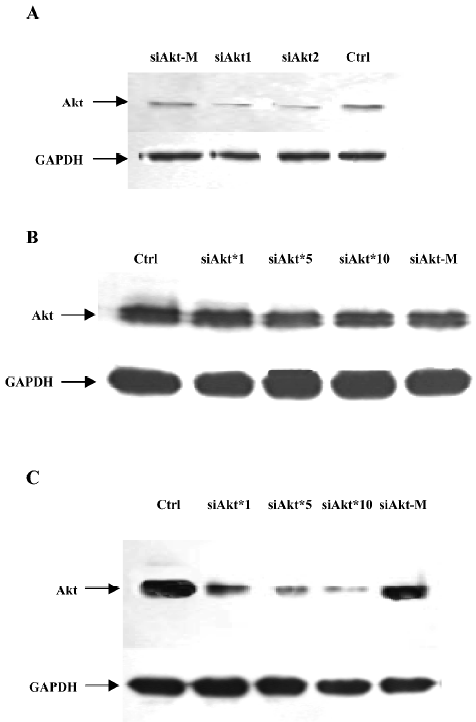
As shown in Figure 3B, in the absence of HCPT, the phagemid particles of siAkt can not efficiently inhibit the Akt expression. However, in the presence of HCPT (Figure 3C), the siAKT phagemid particles can inhibit the Akt expression in a dose-dependent manner. The highest inhibition was 50%−60% when 1×1011 pfu/mL particles were added.
Discussion
In this study, we utilized a novel phagemid system to deliver several siRNA into mammalian cells and estimated their effect in vitro. The phagemid vectors have a number of advantages over other vectors. Unlike other viral gene delivery vector, filamentous phages have no natural mammalian cell tropism. Phagemid particles can be easily retargeted by displaying various ligands, such as peptides, scFv and natural ligands (eg EGF, FGF)[5,8,14–16]. In addition, it is very efficient to prepare the phagemid particles rapidly (about 2–3 d). In previous studies, phages were modified so that they could be used in gene delivery. The ability of cell-targeted phages to deliver siRNA would be of interest. Since phage genomes for packaging replicates by rolling circles, we examined in this study whether the hairpin structure of the siRNA-encoding sequence will impede its replication and package. The results clearly demonstrate that phagemid particles carrying siRNA can be prepared as efficiently as those carrying other protein-encoding genes.
This study also demonstrates that cell-targeted phagemid particles carrying siRNA can be expressed efficiently in the presence of HCPT, although further studies need to be performed to elucidate the mechanism.
We also tested whether the phagemid particles carrying siRNA against Akt have some effect on cell growth. However, no significant inhibition was obtained. This is not uncommon, because the EGF ligand on the phagemid particles has a cell-growth stimulatory effect that might compensate the cell growth inhibitory effect contributed by the siRNA. Thus, to be a cancer gene-delivery vector, phage vectors should display ligands that do not prompt cell growth or carry siRNA against other oncogenes, such as PI3K.
References
- Burg MA, Jensen-Pergakes K, Gonzalez AM, Ravey P, Baird A, Larocca D. Enhanced phagemid particle gene transfer in camptothecin-treated carcinoma cells. Cancer Res 2002;62:977-81.
- Larocca D, Witte A, Johnson W, Pierce GF, Baird A. Targeting bacteriophage to mammalian cell surface receptors for gene delivery. Hum Gene Ther 1998;9:2393-9.
- Larocca D, Kassner PD, Witte A, Ladner RC, Pierce GF, Baird A. Gene transfer to mammalian cells using genetically targeted filamentous bacteriophage. FASEB J 1999;13:727-34.
- Kassner PD, Burg MA, Baird A, Larocca D. Genetic selection of phage engineered for receptor-mediated gene transfer to mammalian cells. Biochem Biophys Res Commun 1999;264:921-8.
- Li ZH, Zhang J, Zhao RJ, Xu YH, Gu JR. Preparation of peptide- targeted phagemid particles using a protein III-modified helper phage. Biotechniques 2005;39:493-7.
- Poul MA, Marks JD. Targeted gene delivery to mammalian cells by filamentous bacteriophage. J Mol Biol 1999;288:203-11.
- Di Giovine M, Salone B, Martina Y, Amati V, Zambruno G, Cundari E, et al. Cell delivery and gene transfer of adenoviral penton base displaying bacteriophage. Virology 2001;282:102-12.
- Li ZH, Jiang H, Zhang J, Shi BZ, Gu JR. Cell targeted phagemid particles preparation using Ecoli bearing ligand-pIII encoding helper phage genome. BioTechniques 2006;41:706-7.
- Sui G, Soohoo C. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 2002;99:5515-20.
- Katome T, Obata T. Use of RNA interference-mediated gene silencing and adenoviral overexpression to elucidate the roles of AKT/protein kinase B isoforms in insulin actions. J Biol Chem 2003;278:28312-23.
- Barbas CF. Phage display: a laboratory manual. CSH Laboratory Press; 2001.
- Rondot S, Koch J, Breitling F, Dübel S. A helper phage to improve single-chain antibody presentation in pahge display. Nat Biotechnol 2001;19:75-78.
- Alexander IE, Russell DW, Miller AD. DNA-damaging agents greatly increase the transduction of nondividing cells by adeno-associated virus vectors. J Virol 1994;68:8282-7.
- Gao CS, Mao SL, Lo CL, Wirsching P, Lerner RA, Janda KD. Making artificial antibodies: a format for phage display of combinatorial heterodimeric arrays. Proc Natl Acad Sci USA 1999;96:6025-30.
- Kwas’nikowski P, Kristensen P, Markiewicz WT. Multivalent display system on filamentous bacteriophage pVII minor coat protein. J Immunol Methods 2005;307:135-43.
- Gao C, Mao S, Ditzel HJ, Farnaes L, Wirsching P, Lerner RA, et al. A cell-penetrating peptide from a novel pVII-pIX phage-displayed random peptide library. Bioorg Med Chem 2002;10:4057-65.
- Zhang R, Li Y, Cai Q, Liu T, Sun H, Chambless B, et al. Preclinical pharmacology of the nature product anticancer agent 10-hydroxycamptothecin, an inhibitor or topoisomerase I. Cancer Chemother Pharmacol 1998;41:257-67.
- Zhou JJ, Liu J, Xu B. Relation between lactone ring forms of HCPT and their antitumor activities. Acta Pharmacol Sin 2001;22:827-30.