
Cellular mechanisms of reduced sarcoplasmic reticulum Ca2+ content in L-thyroxin-induced rat ventricular hypertrophy1
Introduction
The contraction of cardiac myocytes in the heart is initiated when Ca2+ enters the cell via L-type Ca2+ channels in the sarcolemma. Ca2+ entry then triggers the release of a much larger amount of Ca2+ from the sarcoplasmic reticulum (SR)[1,2]. The elementary event of the SR Ca2+ release through ryanodine receptor type 2 (RyR2) is the Ca2+ spark. The spontaneous Ca2+ sparks in quiescent cardiac myocytes reflect the SR Ca2+ content, the function of RyR2, and SR Ca2+-ATPase 2a (SERCA2a), as well as the SR Ca2+ leak[3]. It is the synchronized activation of many Ca2+ sparks triggered by Ca2+ entry via L-type Ca2+ channels that cause the systolic Ca2+ transient and subsequent myocardial contraction[1,2]. There is a re-uptake of released Ca2+ from the SR during contraction into the SR through SERCA2a. Given the dependence of the SR Ca2+ content on the intracellular Ca2+ concentration ([Ca2+]i), SERCA2a function, and spontaneous Ca2+ release, the alteration of the SR Ca2+ content may contribute to abnormal intracellular Ca2+ handling, leading to myocardial dysfunction.
Previous studies have shown that hyperthyroidism causes abnormalities in intracellular Ca2+ signaling com-ponents, which in turn results in cardiac hypertrophy and arrhythmia[4,5]. For example, the enhanced Ca2+ influx through the L-type Ca2+ channel could partly account for the prolonged action potential duration and delayed repolari-zation, and consequently aggravated arrhythmia development during cardiac hypertrophy[6]. The enhanced expression of functional RyR2, increased re-uptake of Ca2+ into the SR through SERCA2a, and decreased phospholamban expression have been reported after thyroxin injection, which was suggested to be responsible, at least in part, for the increase in the SR Ca2+ release and the Ca2+ transient, as well as enhanced myocardial contractility[4,7,8]. In the pressure-overload hypertrophy model, the SR Ca2+ content decreased secondary to the reduced SERCA2a-mediated Ca2+ uptake and increased sarcolemmal-mediated Ca2+ efflux from the cell, which caused the smaller Ca2+ transient and may contribute to the development of arrhythmias during hypertrophy[9]. However, in the thyroxin-induced cardiac hypertrophy model, the change in the SR Ca2+ content and the underlying mechanism have still not been fully understood.
In this study, we used laser scanning confocal microscopy and Ca2+-sensitive fluorescent indicators to examine and quantitatively analyze the SR Ca2+ content, the spontaneous Ca2+ sparks, and the basal [Ca2+]i in quiescent cardiac myocytes from normal rats and L-thyroxin-injected rats with left ventricular hypertrophy (LVH). The lower SR Ca2+ content was identified in this model. The Ca2+ spark recording and analysis demonstrated the increase in the diastolic SR Ca2+ leak, which may be due to more occurrences of spontaneous Ca2+ sparks and an increase in the basal [Ca2+]i. In addition, we observed the decreased activity of SERCA2a, which may lead to the deteriorated function of SERCA2a, contributing to the elevated [Ca2+]i and lower SR Ca2+ content in hypertrophied cardiac myocytes.
Materials and methods
L-thyroxin-induced cardiac hypertrophy All experimental procedures were approved by the Sun Yat-Sen University Committee for Animal Research (Guangzhou, China) and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The L-thyroxin-induced cardiac hypertrophy model was prepared as previously described[4]. Briefly, adult male Sprague–Dawley rats (200±20 g, Experimental Animal Center, Sun Yat-Sen University, China) were randomly divided into 2 groups. The hyperthyroidism group (HT) was injected with L-thyroxin (1 mg/kg, intra-peritoneal) for 10 d to produce hypertrophy. The normal-saline group (NS) was injected with normal saline (controls). The Doppler echocardiographic studies were performed at 10 d to assess the development of heart hypertrophy.
Preparation of cardiac myocytes Single rat ventricular myocytes were isolated from rats using a collagenase-based enzymatic digestion technique[10]. Briefly, the animals were anesthetized with sodium pentobarbital (50 mg/kg, intra-peritoneal). The hearts were quickly removed and perfused in a Langendorff mode. They were first perfused with Ca2+-free Tyrode’s solution composed of (in mmol/L) 136 NaOH, 5.4 KCl, 0.33 NaH2PO4, 1 MgCl2·6H2O, 10 HEPES, and 10 glucose (pH 7.4) at 37 °C for 10 min, then perfused with Ca2+-free Tyrode’s solution containing collagenase (type II, 0.5 mg/mL) for 15 min. The left and right ventricular tissues were removed and myocytes were harvested. The isolated cells were stored in a Krebs–bicarbonate solution containing (in mmol/L) 50 K-glutamate, 20 KOH, 40 KCl, 20 taurine, 20 KH2PO4, 3 MgCl2·6H2O, 10 HEPES, 10 glucose, 0.5 ethylene glycol tetraacetic acid (EGTA), and 1% bovine serum albumin (pH 7.4) at room temperature. This procedure yields 50%–70% of Ca2+-tolerant, rod-shaped ventricular myocytes with clear striations. Cells were used within 10 h after isolation.
Line-scan imaging and Ca2+ spark analysis Myocytes were loaded with 4 µmol/L Fluo-3 AM (Molecular Probes, Eugene, OR, USA) for 30 min at room temperature. The cells were then placed on a Petri plate coated with poly-lysine and were washed for 10 min to allow the de-esterification of the indicator; quiescent myocytes with a typical rod-shaped form and clear cross-striations were used for experiments. Fluo-3 was excited at 488 nm and the Ca2+ fluorescent signal was acquired at 526 nm by confocal microscopy (FV500, Olympus, Tokyo, Japan).
The [Ca2+]i in quiescent cells was reported as fluorescence intensity (FI). To control the background FI, all parameters of confocal microscopy were fixed when different samples were measured. The SR Ca2+ content was assessed by the rapid application of caffeine (20 mmol/L) in Ca2+-free Tyrode’s solution. The amplitude of the caffeine-induced Ca2+ transient could be an index of the SR Ca2+ load[11]. The caffeine-induced Ca2+ transient was derived from changes in FI (F) and normalized to basal fluorescence (F0) and expressed as F/F0.
The spontaneous Ca2+ sparks were captured in Ca2+(1.5 mmol/L)-containing Tyrode’s solution over the entire cell with the confocal microscope operating in x-t imaging mode. The amplitude of spontaneous Ca2+ sparks (F/F0, where F0 refers to the background of the Fluo-3/AM signal), duration (full-duration-half-maximum [FDHM]), width, spatial size (full-width-half-maximum [FWHM]), and Ca2+spark frequency (CaSpF) were measured in a line-scan mode using a 60× water immersion objective by an algorithm coded in IDL 5.4[12] and self-developed program with Matlab 6.5 (Mathworks, Natick, MA, USA). All experiments were performed at room temperature.
Preparation of SR membrane The SR was prepared as previously described[13,14]. Briefly, the isolated ventricle was frozen and homogenated in ice-cold homogenizing medium containing (in mmol/L) 10 NaHCO3 and 5 NaN3, pH 7.0 using Polytron PT-20 (Brinkmann Instruments, Westbury, NY, USA). The homogenate was centrifuged at 14 000×g for 20 min at 4 °C. The pellet was resuspended in 5 volumes of ice-cold buffer and centrifuged as before. The supernatant from the second spin was sedimented at 45 000×g for 30 min and the pellet was resuspended in 25 mL of 0.6 mmol/L KCl and 30 mmol/L histidine, pH 7.0, and centrifuged again. The pellet consisting of the SR was resuspended in the solution containing (in mmol/L) 30 histidine and 250 sucrose, pH 7.4, and was stored at –80 °C.
Preparation of sarcolemma from rat hearts Sarcolemma was prepared from rat ventricles as per the kit manual (Jiancheng, Nanjing, China). Briefly, the left and right ventricles were minced in 9 volumes of ice-cold (0–4 °C) homogenizing medium containing reagent I separately and filtrated by double-deck gauze. The filtrate was sedimented at 10 750×g for 20 min, and the pellet obtained was washed twice by reagent I. The pellet was then suspended with 10 mL reagent II and placed at 0 ºC. After 48 h, the sediment was centrifuged for 20 min at 10 750×g and then washed twice by reagent III and preserved in reagent IV at 0 °C; the activity of Ca2+-ATPase was measured within 48 h.
Measurement of Ca2+-ATPase activity The activity of Ca2+-ATPase was determined as per the kit manual (Jiancheng, China) by measuring the inorganic phosphate liberated from ATP hydrolysis[14]. Briefly, Ca2+-ATPase activity was assayed in a medium containing (in mmol/L) 50 histidine, 3 MgCl2, 100 KCl, 5 sodium azide, 3 ATP, and 0.05 CaCl2, pH 7.0. The cardiac SR membrane was added to the reaction mixture at a final concentration of 25 µg of protein/mL, pre-incubated for 10 min at 37 °C, and the reaction was initiated by the addition of ATP. The ATP hydrolysis that occurred in the absence of Ca2+ (1 mmol/L EGTA) was subtracted to determine the activity of Ca2+-stimulated ATPase. Ouabain was added freshly to a final concentration of 1 mmol/L in the media, which remained unchanged throughout the incubation. Mitochondrial contamination was excluded by determining the activity of azide-sensitive ATPase[15].
Statistics All data were expressed as mean±SEM. The differences between the groups were analyzed by paired t-test or ANOVA, P<0.05 was considered significant.
Results
L-thyroxin injection created LVH All of the rats were killed 10 d after the injection to examine the gross indexes of hypertrophy. Compared with normal controls, the ratios of heart weight to brain weight (HW/BW) and left ventricular weight to BW (LVW/BW) in the HT group were increased significantly by ≈20% and ≈33%, respectively, whereas the ratio of right ventricular weight to BW (RVW/BW) showed no significant difference between the control and HT rats (Table 1). Doppler echocardiography demonstrated that the HT group had increased interventricular septum end-diastolic thickness (IVSd) and interventricular septum end-systolic thickness (IVSs; P<0.05; Table 1), but left ventricle end-diastolic dimension (LVDd), left ventricle end-systolic dimension (LVDs), end-diastolic posterior wall thickness (PWd), and end-systolic posterior wall thickness (PWs) were normal and similar in the HT and NS groups. Left systolic ventricular function was assessed by ejection fraction (EF) and left ventricular fractional shortening (LVFS); both were not significantly different from the NS group (P>0.05; Table 1).The increased LVW/BW and preserved left ventricular function in the HT group suggested that the exposure to L-thyroxin produced compensated LVH.
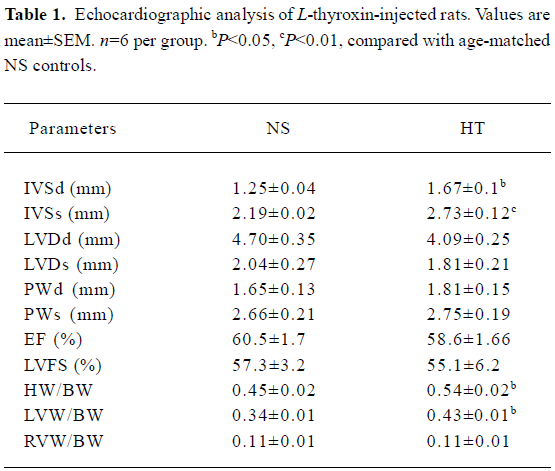
Full table
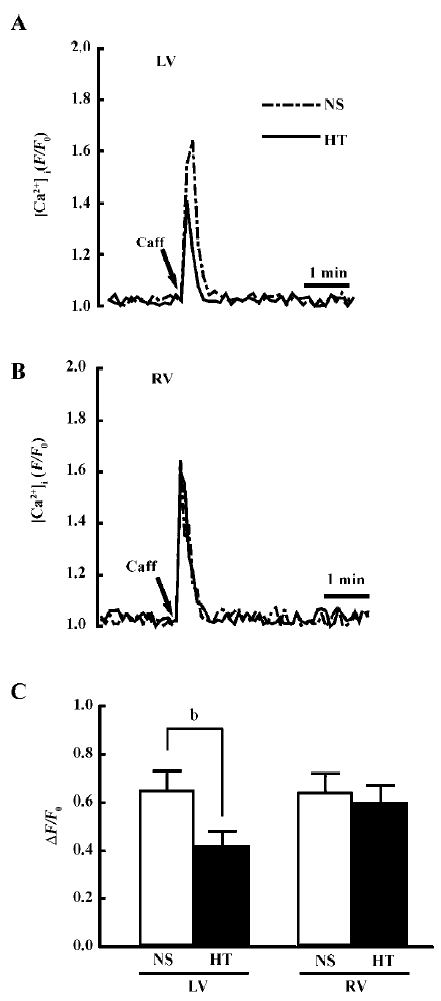
SR Ca2+ content In the Ca2+-free medium, the difference in the caffeine-induced Ca2+ transient reflects the change in the SR Ca2+ content. Figure 1A and 1B shows the representative recordings of the Ca2+ transient upon the application of 20 mmol/L caffeine in the left and right ventricular myocytes from the NS and HT groups, Figure 1C compares the amplitude of the caffeine-induced Ca2+ transient (ΔF/F0) between different groups. In the left ventricular myocytes, the mean values of ΔF/F0 in the HT group was significantly lower than in the NS (0.42±0.06, n=21 vs 0.65±0.08, n=27; P<0.05). However, the regional difference in the SR Ca2+ content was absent in right ventricular myocytes.
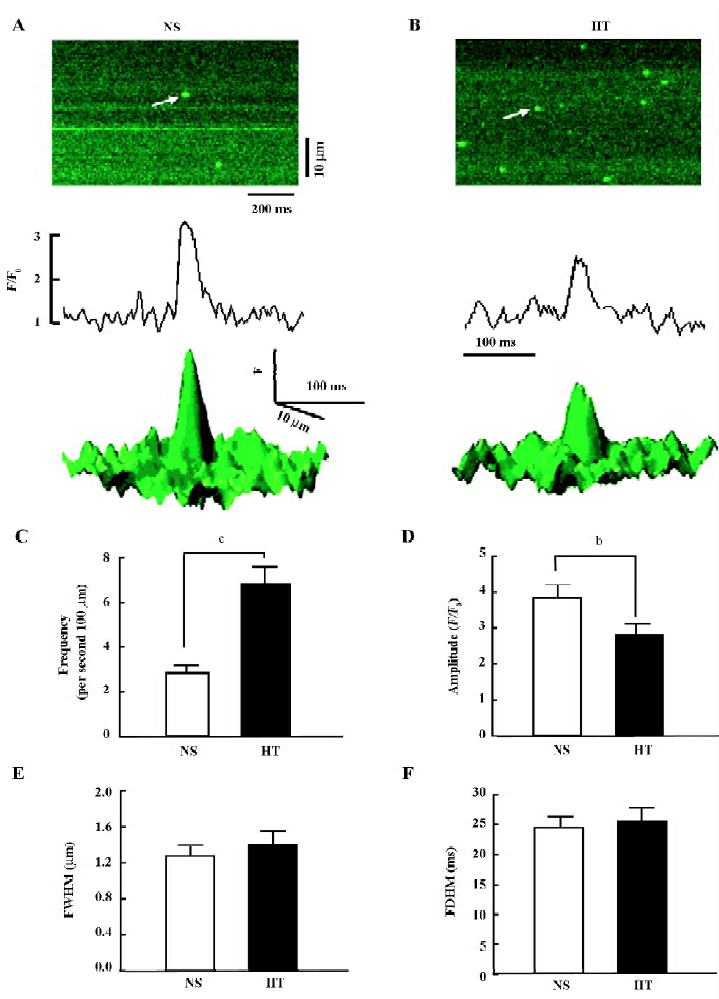
Spontaneous Ca2+ sparks Confocal microscopy was applied to directly quantify the Ca2+ spark (Figure 2A,2B). The Ca2+ spark is the basic Ca2+ release event from the SR, and it is a local, discrete elevation in myoplasmic [Ca2+]i due to the opening of the RyR2[18]. Figure 2C–2F summarizes the characteristics of spontaneous Ca2+ sparks in the NS and HT groups. The CaSpF was higher in the HT animals than in the NS animals (Figure 2C; 6.86±0.74 vs 2.89±0.32 sparks/s*100 µm; P<0.01). The mean amplitude of the Ca2+ spark was lower in HT group than in NS group (Figure 2D; 2.84±0.28 vs 3.87±0.34, F/F0; P<0.01), consistent with the lower SR Ca2+ content. The width and duration of the Ca2+ sparks were not significantly changed in the HT group compared with those in the NS group (Figure 2E,2F; FWHM: 1.4±0.15 vs 1.29±0.1 μm; FDHM: 25.7±2.14 vs 24.6±1.8 ms; P>0.05). The diastolic SR Ca2+ leak was found to be related to the product CaSpF×amplitude×FDHM×FWHM[16], which was 1.5 times higher in the HT group than the NS group.
Because of the decreased SR content and increased diastolic SR Ca2+ leak, we further compared the basal [Ca2+]i between the NS and HT groups. The basal [Ca2+]i was significantly elevated in quiescent left ventricular myocytes from the HT group compared with that from the control group (1432±153, n=38 vs 1143±144, n=32; P<0.05), whereas the basal [Ca2+]i was unchanged in the right ventricle myocytes from the hypertrophied heart (1110±123, n=40 vs 1150±130, n=63; P >0.05).
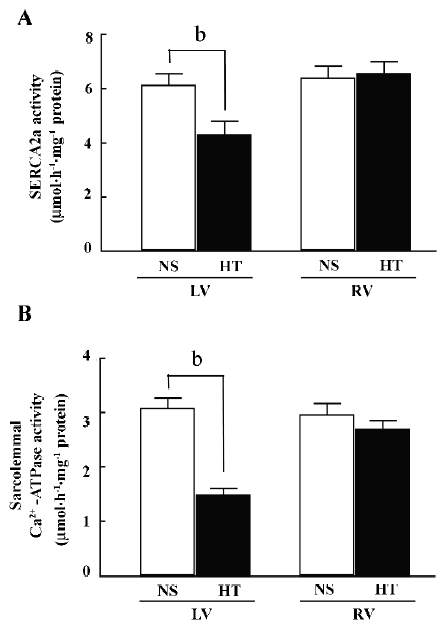
Ca2+-ATPase activity SERCA2a is the key Ca2+-transport protein that re-uptakes Ca2+ into the SR during relaxation. The activity of SERCA2a in left ventricular myocytes from the HT group was significantly lower than in the NS group (4.34±0.44 vs 6.15±0.41 µmol·h–1·mg–1 protein, n=6, P<0.01; Figure 3A), whereas there was no obvious change in the right ventricle in both groups (n=6, P>0.05). The activity of sarcolemmal Ca2+-ATPase in the hypertrophied left ventricle decreased significantly compared with the NS controls (1.49±0.12 vs 3.09±0.18 µmol·h–1·mg–1 protein, n=6, P<0.01; Figure 3B), whereas there was no obvious change in the right ventricle (n=6, P>0.05).
Discussion
In the present study, it was demonstrated that the ventricular myocytes from the L-thyroxin-induced hypertrophy model decreased the caffeine-induced Ca2+ transient in the Ca2+-free solution. The smaller caffeine-induced Ca2+ transient could be explained by the lower SR Ca 2+ content. We also observed the increased Ca2+ leak, reduced SERCA activity, and increased basal [Ca2+]i. in hypertrophied ventricular myocytes in hyperthyroidism rats, which may be involved in the possible mechanisms for the lower SR Ca2+ content in ventricular myocytes in this hypertrophy model.
The SR Ca2+ content in cardiac cells reflects the balance between Ca2+ release through RyR and Ca2+ uptake into the SR via SERCA2a. The basic Ca2+ release event was quantitatively detected and analyzed by a Ca2+ spark recording and analysis. Since the SR Ca2+ content mostly consists of the Ca2+ transient (approximately 92%) leading to the initiation of extracellular Ca2+ entry and subsequent myocardial contraction in adult hearts[17], the decrease in the amplitude of Ca2+ sparks indicates the lower SR Ca2+ content.
Previous studies have shown the frequency of Ca2+ sparks increases as the SR Ca2+ load elevates, and the smaller SR Ca2+ content is accompanied by fewer Ca2+ spark rates[18,19]. However, we observed a higher Ca2+ spark frequency with a decrease in SR Ca2+ content in this hypertrophy model. The paradoxical observation was also found in a severe but compensated canine LVH model, where the hyperphosphorylation of RyR2 was demonstrated to cause pathological hypersensitivity of RyR2 and release more Ca2+ in the diastolic period, leading to an increased occurrence of Ca2+ spark frequency[3]. In the same L-thyroxin-induced hypertrophy model, the increased expression of RyR2 has been reported in the hypertrophied heart tissue[4,7]. Therefore, more RyR2 in the SR and/or its hypersensitivity may be able to evoke more Ca2+ sparks. Another possibility for increased occurrences of spontaneous Ca2+ sparks may be due to the firing of Ca2+ release from some “Ca2+-overloaded” subcellular regions in hypertrophied myocytes[3]. However, this possibility requires further examination in the L-thyroxin induced hypertrophy model.
The spontaneous Ca2+ leak occurs as the loss of Ca2+ from the SR under resting conditions, which also plays a role in the diastolic removal of Ca2+ from the SR Ca2+ content[20]. The enhanced SR Ca2+ leak was reported in the hypertrophy model induced by the Calcium/calmodulin-dependent protein kinase type II delta (CaMKIIδ) overexpression[16]. This model also points out that the enhanced expression of RyR2 with hypersensitivity may contribute to the increased SR Ca2+ leak. The increased Ca2+ leak and higher basal [Ca2+]i that we observed in the present study may therefore explain the arrhythmogenesis in the hyperthyroid heart. In addition, the SR Ca2+ release channels are activated as [Ca2+]i elevates. Therefore, the increased Ca2+ spark frequency during hypertrophy may also be secondary to an increase in [Ca2+]i.
The size of the SR Ca2+ content is dependent on the Ca2+ re-uptake through SERCA2a. The smaller SR Ca2+ content may be associated with reduced SERCA2a function. In the L-thyroxin-induced cardiac hypertrophy model, enhanced RyR2 and the SERCA2a mRNA level was observed and was associated with Ca2+ overload contributing to arrhythmogenesis during hypertrophy[4]. The expression of SERCA2a RNA and protein has been observed in hypertrophy models[4,21], but whether the activity of SERCA2a was altered had not previously been examined. In the present study, we observed a marked decrease in the activity of SERCA2a in the hypertrophied heart, which seemed to result in decreased SERCA2a function and may have contributed to the decreased SR Ca2+ content and increased basal [Ca2+]i. However, another possibility could not be excluded. The augmented expression level with the decreased activity of SERCA2a may not only cause more Ca2+ re-uptake back into Ca2+ store, but also cause oxygen wastage, which is consistent with increased oxygen consumption in the hyperthyroid heart.
In addition, there is controversy over whether there is change in the SERCA2a expression in the hyperthyroid heart. Takeuchi et al reported that there was no change in the SERCA2a protein expression in the hyperthyroid heart and proposed that the SERCA2a activity would be enhanced to lead to metabolic derangement[22], but they did not examine the SERCA2a activity in their study. It should be noted that the activity of ATPase (Na+/K+-ATPase and K+/Ca2+-ATPase) at the sarcolemma, SR, and mitochondria might be differently modified in the process of hypertrophy[23]. Because we facilitated the procedure to examine the activity of ATPase in subcelluar populations enriched in the SR and sarcolemma, respectively, the possible contamination from mitochondria was excluded. Although we did not further examine how SERCA2a function is altered and how myocardial contraction changes, the present study raises decreased SERCA2a activity as a potential mechanism for decreased SR Ca2+ content.
In the present study, we found that the elevated basal [Ca2+]i could be due to the increased Ca2+ leak and reduced SERCA2a activity. In diastolic Ca2+ removal from the cytosol in the rat heart, the contribution of SERCA2a has been demonstrated to be predominant than that of the Na+–Ca2+ exchange[24]. However, it is noteworthy that acute exposure to the thyroid hormone stimulated the activity of reverse mode Na+–Ca2+ exchange in cat atrial myocytes and increased [Ca2+]i, which was suggested to be involved in Ca2+-mediated arrhythmic activity[25]. Because this mode of Ca2+ influx may also account for the increased basal [Ca2+]i in the hyperthyroid ventricle[25], the role of Na+–Ca2+ exchange in rat hypertrophied ventricular myocytes needs further investigation.
In summary, the results of our present study suggest that the increased Ca2+ leak and reduced SERCA2a activity may contribute to decreased SR Ca2+ content and increased basal [Ca2+]i in ventricular myocytes in the L-thyroxin-induced hypertrophy model.
Acknowledgments
We thank Prof Jian-xin SHEN (Department of Physiology, University of Shan Tou) and Yan-qiu FENG (Department of Biomedical Engineering, The Southern Medical University) for their help with the Ca2+ spark recording.
References
- Guatimosim S, Dilly K, Santana LF, Saleet Jafri M, Sobie EA, Lederer WJ. Local Ca2+ signaling and EC coupling in heart: Ca(2+) sparks and the regulation of the [Ca2+]i transient. J Mol Cell Cardiol 2002;34:941-50.
- Balke CW, Shorofsky SR. Alterations in calcium handling in cardiac hypertrophy and heart failure. Cardiovasc Res 1998;37:290-9.
- Song LS, Pi Y, Kim SJ, Yatani A, Guatimosim S, Kudej RK, et al. Paradoxical cellular Ca2+ signaling in severe but compensated canine left ventricular hypertrophy. Circ Res 2005;97:457-64.
- Wu XD, Dai DZ, Zhang QP, Gao F. Propranolol and verapamil inhibit mRNA expression of RyR2 and SERCA in L-thyroxin-induced rat ventricular hypertrophy. Acta Pharmacol Sin 2004;25:347-51.
- Dillmann WH. Cellular action of thyroid hormone on the heart. Thyroid 2002;12:447-52.
- Dai DZ, Hu HJ, Yang DM, Hao XM, Zhang GQ, Zhou PA, et al. Chronic levothyroxin treatment is associated with ion channel abnormalities in cardiac and neuronal cells. Clin Exp Pharmacol Physiol 1999;26:819-21.
- Jiang M, Xu A, Tokmakejian S, Narayanan N. Thyroid hormone-induced overexpression of functional ryanodine receptors in the rabbit heart. Am J Physiol Heart Circ Physiol 2000;278:H1429-38.
- Shenoy R, Klein I, Ojamaa K. Differential regulation of SR calcium transporters by thyroid hormone in rat atria and ventricles. Am J Physiol Heart Circ Physiol 2001;281:H1690-6.
- Diaz ME, Graham HK, Trafford AW. Enhanced sarcolemmal Ca2+ efflux reduces sarcoplasmic reticulum Ca2+ content and systolic Ca2+ in cardiac hypertrophy. Cardiovasc Res 2004;62:538-47.
- Xiao RP, Ji X, Lakatta EG. Functional coupling of the beta 2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol 1995;47:322-9.
- Negretti N, O’Neill SC, Eisner DA. The relative contributions of different intracellular and sarcolemmal systems to relaxation in rat ventricular myocytes. Cardiovasc Res 1993;27:1826-30.
- Cheng H, Song LS, Shirokova N, Gonzalez A, Lakatta EG, Rios E, et al. Amplitude distribution of calcium sparks in confocal images: theory and studies with an automatic detection method. Biophys J 1999;76:606-17.
- Jones LR, Besch HR Jr, Fleming JW, McConnaughey MM, Watanabe AM. Separation of vesicles of cardiac sarcolemma from vesicles of cardiac sarcoplasmic reticulum. Comparative biochemical analysis of component activities. J Biol Chem 1979;254:530-9.
- Kodavanti PR, Cameron JA, Yallapragada PR, Desaiah D. Effect of chlordecone (Kepone) on calcium transport mechanisms in rat heart sarcoplasmic reticulum. Pharmacol Toxicol 1990;67:227-34.
- Lindemann JP, Jones LR, Hathaway DR, Henry BG, Watanabe AM. α-Adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J Biol Chem 1983;258:464-71.
- Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res 2003;92:904-11.
- Delbridge LM, Satoh H, Yuan W, Bassani JW, Qi M, Ginsburg KS, et al. Cardiac myocyte volume, Ca2+ fluxes, and sarcoplasmic reticulum loading in pressure-overload hypertrophy. Am J Physiol 1997;272:H2425-35.
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 1993;262:740-44.
- Santana LF, Cheng H, Gomez AM, Cannell MB, Lederer WJ. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ Res 1996;78:166-71.
- Sobie EA, Guatimosim S, Gomez-Viquez L, Song LS, Hartmann H, Saleet Jafri M, et al. The Ca2+ leak paradox and rogue ryanodine receptors: SR Ca2+ efflux theory and practice. Prog Biophys Mol Biol 2006;90:172-85.
- Kim YK, Kim SJ, Yatani A, Huang Y, Castelli G, Vatner DE, . Mechanism of enhanced cardiac function in mice with hypertrophy induced by overexpressed Akt. J Biol Chem.2003; 278: 47 622–8.
- Takeuchi K, Minakawa M, Otaki M, Odagiri S, Itoh K, Murakami A, et al. Hyperthyroidism causes mechanical insufficiency of myocardium with possibly increased SR Ca2+-ATPase activity. Jpn J Physiol 2003;53:411-6.
- Moisin C, Balta N, Filcescu V, Dumitriu IF, Stoian G, Petec G. Activity of Na+/K+-ATPase and of Ca++-ATPase under the action of adenosine triphosphate in experimental myocardial hypertrophy. Rom J Physiol 1998;35:303-11.
- Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol 1994;476:279-93.
- Wang HL, Dai DZ, Gao E, Zhang YP, Lu F. Dispersion of ventricular mRNA of RyR2 and SERCA2 associated with arrhythmogenesis in rats. Acta Pharmacol Sin 2004;25:738-43.