
Protective effects of TREK-1 against oxidative injury induced by SNP and H2O21
Introduction
Two-pore-domain K+ channels (K2P), a novel class of K+ channels, are responsible for background K+ currents in various cells[1–3]. These channels are open at membrane potentials within the physiological range, so they are thought to mediate the background or leak currents. TWIK-related potassium channel (TREK) is a subfamily of K2P and consists of 3 identified channel proteins: TREK-1, TREK-2, and TRAAK (TWIK-related arachidonic acid-stimulated K+ channel)[3,4].
TREK-1 was the first mammalian mechano-gated ion channel to be cloned, expressed, and characterized[5,6]. Human TREK-1 is a 411 amino acid polypeptide and its mRNA is mainly expressed in various regions of the brain[6–8]. TREK-1 possesses unique properties that may have important physiological and pathophysiological significance. These include their sensitivity to unsaturated free fatty acids, lysophospholipids, pressure or membrane stretch, intracellular pH, oxygen tension, heat, and neuro-transmitters that are coupled to G-proteins[9-11]. Recent in vivo studies have indicated that TREK-1 plays a key role in the cellular mechanisms of neuroprotection, anesthesia, pain, and depression[12,13].
Oxidative stress and free radical damage to tissues may be involved in the development of diseases, such as cancer, neurodegenerative diseases, and cardiovascular diseases[14–17]. As a nitric oxide (NO)-generating agent, sodium nitroprusside (SNP) generates NO when added to cell culture. When NO levels exceed physiological levels by continued NO generation and accumulation, NO may initiate a toxic cascade and lead to cell death[17]. Studies have suggested that the toxic action of NO may also be relevant to the pathogenesis of neurodegenerative disease such as Alzheimer’s disease[18,19]. Hydrogen peroxide (H2O2), a non-radical molecule that generates highly toxic hydroxyl radicals via the Fenton reaction, could also be involved in cell death and various disease processes[20].
In this study, we compared the cell viability and apoptotic rate in TREK-1/Chinese hamster ovary (CHO) cells with those of wild-type CHO cells to examine the role of TREK-1 in the process of oxidative stress and apoptosis induced by SNP/H2O2.
Materials and methods
Cell culture and stable transfection CHO cells were maintained in a 95% air/5% CO2 (v/v) humidified atmosphere at 37 °C. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 U/mL), and 2 mol/L glutamine. cDNA encoding rTREK-1 channel (GenBank Accession No. AY695826) cloned into a pcDNA mammalian expression vector was stably transfected into CHO cells using Lipofectamine 2000 reagent. In brief, the CHO cells were transfected when they were at 20%–30% confluency using Lipofectamine 2000. DNA (2 µg) was diluted in DMEM (250 µL) and mixed with Lipofectamine 2000 (10 µL). Complexes were allowed to form for 20 min at room temperature before being added to the CHO cells in a 35-mm dish. After 24–48 h of transfection, the cells were split and seeded at low density for G418 selection. The selection medium (G418, 800 mg/L) was replaced every 5 d. After 2–3 weeks, G418-resistant colonies were selected. An rTREK-1 expressing clone was then identified by using whole-cell electrophysiology. This positive clone was passaged every 3 d by trypsin treatment and for regular experiments.
Electrophysiology Whole-cell patch clamp recordings were performed using a List EPC-9 amplifier (HEKA Elektronic, Lambrecht, Germany). Pipettes with tip resistance of 3–5 MΩ were used. Under the current clamp mode, membrane potentials were measured when cells were held at 0 mA. To evoke whole-cell K+ currents, cells were held at –80 mV with a 500 ms pulse from –80 mV to +80 mV. The external solution contained in mmol/L: 135 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, and 5 HEPES, with the pH adjusted to 7.4 with NaOH. The patch pipette solution contained in mmol/L: 160 KCl, 0.5 MgCl2, and 10 HEPES, pH 7.2 with KOH. Data were stored in an IBM computer by using Pulse software (HEKA Elektronic, Lambrecht, Germany). Series resistance was in the range of 6–12 MΩ and was compensated by 80%. All experiments were performed at room temperature (21–24 °C).
Oxidative injury To induce oxidative stress, 2 free radical generating systems were carried out, including SNP and H2O2. SNP was applied as a NO donor, while H2O2 was used as a precursor of highly reactive free radicals. The cells were seeded in a 96-well plate at a density of 1×104 cells/mL, and 200 µL cell suspension was added to each well. When cells reached 60%–70% confluency, the culture medium (DMEM+FBS) was replaced by DMEM (containing glucose) for a further 24 h before exposure to SNP or H2O2. SNP and H2O2 were prepared in phosphate-buffered saline (PBS) to desirable concentrations just before use. For the SNP-treated cells, SNP was added to the cells with final concentrations of 0.1, 0.25, 0.5, 0.75, 1, 2.5, and 5 mmol/L, respectively. The control cells received identical treatment, although SNP was omitted from the PBS. For the H2O2-treated cells, 0.01, 0.05, 0.1, 0.2, or 0.5 mmol/L H2O2 in PBS was used.
Cell viability assay Cell viability was determined with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay[21]. Briefly, the cells were incubated with SNP or H2O2 in the culture medium for 6, 12, and 24 h and then with 50 g/L MTT for another 4 h. The optical density was assessed at 570 nm using an ELISA plate reader (Molecular Devices, Sunnyvale, CA, USA) to determine the number of viable cells after oxidative injury. The cell viability was expressed as a percentage of the DMSO-treated control samples. All data presented in our study were obtained from at least 3 independent experiments.
Identification of apoptosis by Hoechst33342 staining Morphological changes that are characteristic of apoptosis in the cells were studied by Hoechst33342 staining[22]. The cells were seeded in 96-well plates and treated with 0.75 mmol/L SNP or 0.05 mmol/L H2O2 for 12 h, respectively. After that, Hoechst33342 (10 mg/L) was added to the cells and incubated for 30 min at 37 °C in the dark. The cells were then fixed with 4% paraformaldehyde for 10 min at room temperature, counted, and examined by fluorescence microscopy at 480 nm. Apoptotic cells were identified by their characteristic nuclei condensation and fragmentation, whereas nuclei from normal cells had a normal uniform chromatin pattern. The percentage of apoptotic cells was calculated by the ratio of apoptotic cells to the total cells counted. At least 500 cells were counted from more than 3 random microscopic fields.
Materials SNP, H2O2, Hoechst 33342, and arachidonic acid (AA) were purchased from Sigma–Aldrich (St Louis, MO, USA). DMEM, FBS, G418, trypsin, MTT, and Lipofectamine 2000 reagent were purchased from Gibco (Grand Island, NY, USA). Other medicals reagents were from Beijing Chemical Factory (Beijing, China).
Statistical analysis Data were expressed as mean±SEM. Statistical analysis was performed by one-way ANOVA followed by least significant difference (LSD) comparison test or by paired Student’s t-test. Statistical significance was considered at P<0.05.
Results
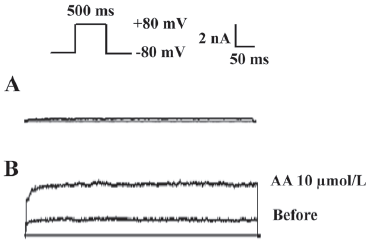
Electrophysiological properties of TREK-1/CHO cells Under voltage clamp, a depolarizing step to +80 mV from the holding potential of –80 mV evoked an outward current in wild-type CHO with a mean amplitude of 48.4±35.6 pA (Figure 1A; n=5). In contrast, the same voltage protocol elicited a significantly larger outward current (1.8±0.6 nA; n=8, P<0.05) in cells expressing TREK-1 (Figure 1B). Consistent with previous reports, this current was significantly enhanced by 10 µmol/L AA (Figure 1B; n=5, P<0.05)[5,10]. Because the current elicited in transfected cells was significantly larger than that of the wild-type cells, we considered this current to be passed by the TREK-1 K+ channels. The outward current elicited reached a steady state without delay. The membrane potential (Em) in the TREK-1/CHO cells was –54.2±1.4 mV (n=8), while the Em in wild-type CHO cells was –23.5±2.4 mV (n=7; data not shown).
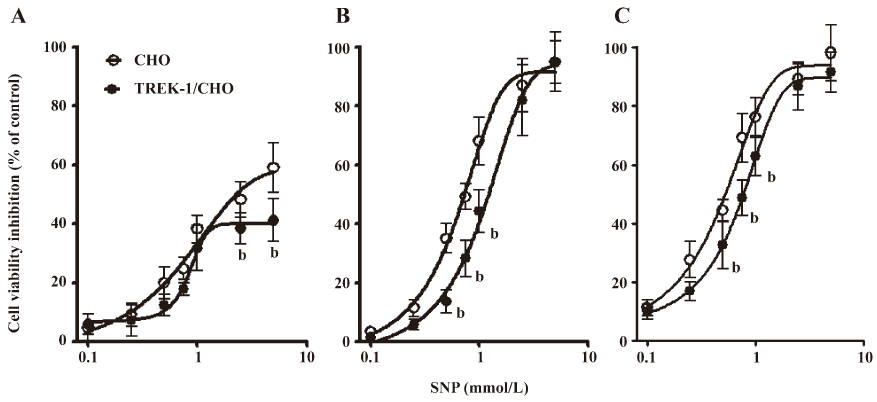
SNP-induced cell death in wild-type CHO cells and TREK-1/CHO cells The effects of SNP and H2O2 on cell viability were examined both in wild-type CHO and TREK-1/CHO cells. As shown in Figure 2, SNP inhibited growth in both cell types in a dose- and time-dependent manner. For TREK-1/CHO cells, the addition of SNP at concentrations of 0.1–5 mmol/L to cell cultures for 6, 12, and 24 h resulted in a significant dose-dependent decrease in the number of viable cells. The effective SNP concentration for 50% inhibition (IC50) was calculated to be 1.36, 1.14, and 0.78 mmol/L for 6, 12, and 24 h exposure, respectively. Compared with the TREK-1/CHO cells, SNP (0.1–5 mmol/L) exposure for 6, 12, and 24 h decreased the viable cells (IC50: 0.85, 0.69, and 0.55 mmol/L, respectively) in wild-type CHO cells. TREK-1/CHO cells were more resistant to SNP (0.1–5 mmol/L) injury than wild-type CHO cells within 24 h (Figure 2; P<0.05).
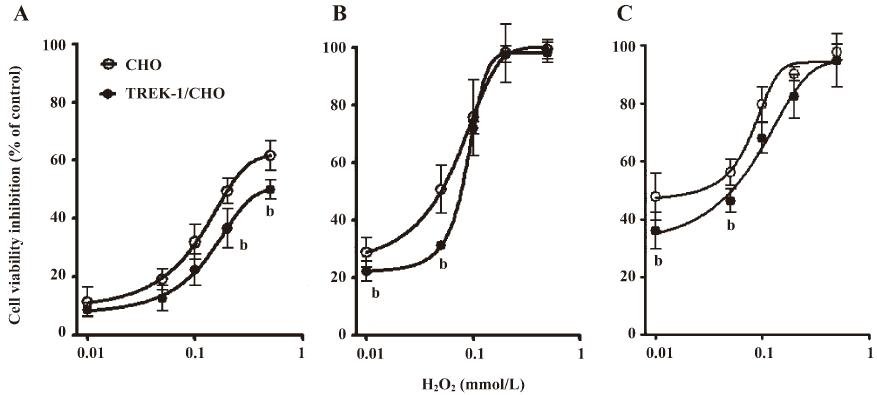
H2O2-induced cell death in wild-type CHO and TREK-1/CHO cells H2O2 treatment inhibited cell growth in a time- and dose-dependent manner by exposure for 6–24 h (Figure 3). When treated for 12 h, the IC50 of H2O2 was 0.07 mmol/L for wild-type CHO cells, and 0.09 mmol/L for TREK-1/CHO cells. Several concentrations of H2O2 (0.01 and 0.05 mmol/L) resulted in significant differences in cell viability between wild-type CHO cells and TREK-1/CHO cells (P<0.05; Figure 3B). The IC50 was calculated as 0.13 and 0.08 mmol/L for wild-type CHO cells, and 0.16 and 0.10 mmol/L for TREK-1/CHO cells 6 and 24 h after exposure to H2O2, respectively. Similar to SNP treatment, the dose-response curves of 6, 12, and 24 h exposure to H2O2 showed that wild-type CHO cell growth was inhibited more than TREK-1/CHO cell growth (Figure 3; P<0.05).
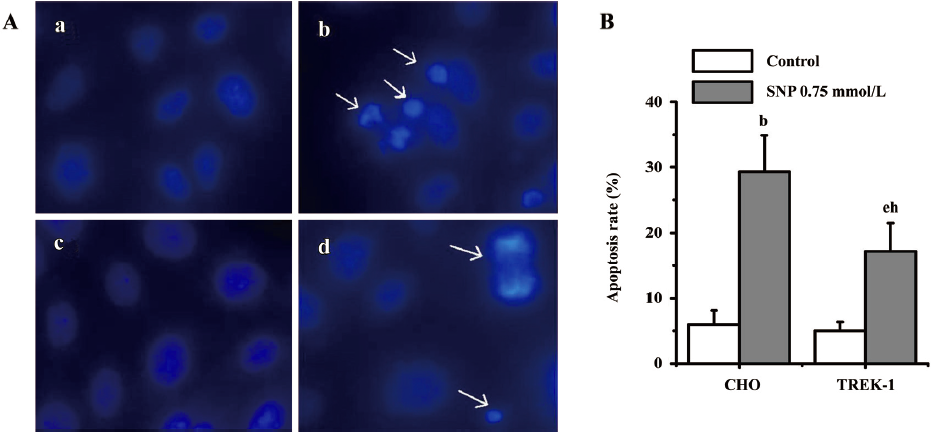
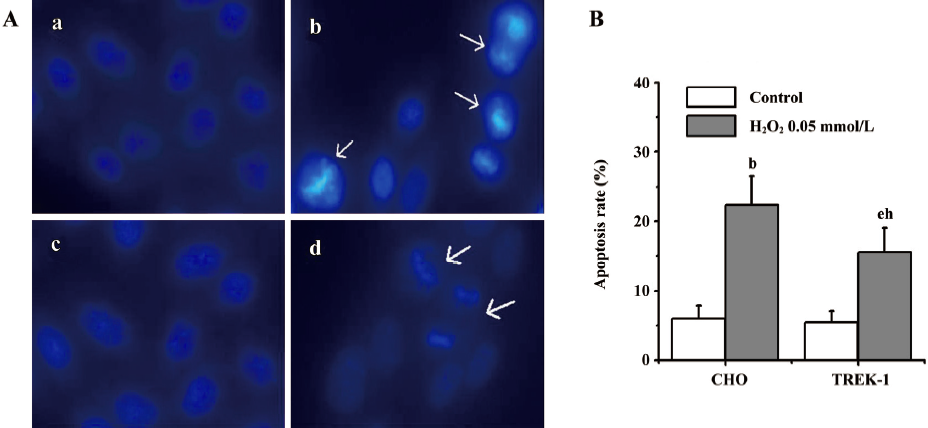
Effects of TREK-1 on SNP/H2O2-induced apoptosis The Hoechst33342 staining technique was used to investigate whether SNP/H2O2 induced apoptosis in both cell types. Following treatment with 0.75 mmol/L SNP for 12 h, both wild-type CHO and TREK-1/CHO cells showed obvious nuclear condensation, which is typical of apoptotic morphology. In contrast, most of the control cells (wild-type CHO or TREK-1 /CHO cells without any treatment) had intact nuclei (Figure 4A). Approximately 29.3%±2.4% of CHO cells showed DNA changes when subjected to SNP, which was significantly greater than the untreated control cells (6.2%±1.1%; P<0.05). When treated with 0.75 mmol/L SNP, 17.3%±3.2% of TREK-1/CHO cells appeared apoptotic, whereas only 5.0%±1.3% of untreated control cells were apoptotic. Apoptosis in CHO cells was significantly greater than that in TREK-1/CHO cells following 12 h exposure to SNP (Figure 4B; P<0.05). Similar to SNP treatment, 0.05 mmol/L H2O2 treatment in both TREK-1/CHO and wild-type CHO cells resulted in condensed chromatin in many cells (Figure 5A). DNA changes occurred in 22.3%±3.0% of H2O2-treated CHO cells, and 15.6%±1.2% of TREK-1/CHO cells were apoptotic when exposed to H2O2 for 12 h. H2O2 induced greater apoptosis in wild-type CHO cells than that in TREK-1 /CHO cells (Figure 5B; P<0.05).
Discussion
In this study, we found that TREK-1/CHO cells had greater viability than wild-type CHO cells when subjected to SNP/H2O2 at equivalent doses within 24 h. These results suggested that TREK-1 transfected cells were more tolerant to oxidative stress induced by SNP/H2O2 than wild-type CHO cells, which indicated that the overexpression of TREK-1 might protect CHO cells against oxidative stress induced by SNP/H2O2.
Previous studies showed that the exposure of CHO cells to SNP/H2O2 resulted in apoptosis[23,24]. In our study, we observed apoptosis in TREK-1/CHO cells and found that apoptosis occurred more in wild-type CHO cells than in TREK-1/CHO cells when subjected to the same concentrations of SNP/H2O2.
In the present study, dose-response curves measured 6 h after exposure to SNP/H2O2 showed that the maximum inhibition of cell viability was less than 65% for TREK-1/CHO cells, and less than 55% for wild-type CHO cells. We hypothesize that these findings might reflect the duration of exposure being too short to trigger apoptosis, thus little serious damage could occur in the 2 cell types. Following 12 or 24 h exposure to SNP/H2O2, the maximum inhibition of cell viability reached nearly 100%. In these cases, cell death may result from apoptosis as well as other cell death pathways.
The TREK-1 channel is expressed in various tissues and cell types, such as hippocampal neurons and cardiomyocytes, and these channels mediate background K+ current[3]. Unfortunately, no specific blockers for TREK-1 are currently available[3]. In this study, we transfected CHO cells with TREK-1 channel DNA in order to study the properties of TREK-1 as well as to screen compounds which specifically act on the TREK-1 channel. We found that the membrane potential of TREK-1/CHO cells was more negative than that of wild-type CHO cells, indicating that the overexpression of TREK-1 could shift the cell membrane potential to the hyperpolarization direction, which may have a protective effect.
Intracellular K+ is an important component that sta-bilizes ion stasis and maintains cellular volume. The effects of K+ efflux on cell injury/cell protection have been discussed extensively[25–30]. Previous studies revealed that delayed rectifying K+ currents increased in the early stage of apoptosis in hippocampal neurons and cortical neurons. Moreover, the K+ channel blocker, tetraethylammonium (TEA), could inhibit apoptosis[28,31]. The overexpression of DNA for the voltage-gated delayed rectifying K+ channel Kv2.1 has been shown to increase CHO cell sensitivity to apoptotic inducers[27]. These results showed that K+ efflux could promote apoptosis. However, efflux of K+ also has a protective effect by hyperpolarizing the membrane as well as decreasing the membrane excitability and oxygen consumption. Membrane hyperpolarization following the activation of K+ channels increased the tolerability of murine myoblasts from apoptosis[29]. Lee et al found that the overexpression of small conductance, calcium-activated K+ channel and the voltage-gated K+ channel Kv1.1 in rat hippocampal slices reduced apoptotic cell death by protective hyperpolarization[31]. Our data indicated that the overexpression of TREK-1 might protect the cells against apoptosis-inducing stress by a similar mechanism.
The present study demonstrates the protective role of TREK-1 against oxidative stress induced by SNP/H2O2. Membrane hyperpolarization was observed after the overexpression of TREK-1 in CHO cells, which suggested that the protective effects of TREK-1 might be mediated by cell hyperpolarization, cell damage reduction, and the inhibition of apoptosis. Further studies should investigate the molecular mechanisms of TREK-1 in oxidative stress, including channel regulation and the signal transduction pathway between TREK-1 and apoptosis.
References
- Goldstein SA, Price LA, Rosenthal DN, Pausch MH. ORK1, a potassium selective leak channel with two pore domains cloned from by expression in . Proc Natl Acad Sci USA 1996; 93: 13 256–61.
- Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, et al. TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J 1996;15:1004-11.
- Patel AJ, Honorè E. Properties and modulation of mammalian 2P domain K+ channels. Trend in Neurosci 2001;24:339-46.
- Gutman GA, Chandy KG, Adelman JP, Aiyar J, Bayliss DA, Clapham DE, et al. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol Rev 2003;55:583-6.
- Patel AJ, Honoré E, Maingret F, Lesage F, Fink M, Duprat F, et al. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J 1998;17:4283-90.
- Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, et al. Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J 1996;15:6854-62.
- Medhurst AD, Rennie G, Chapman CG, Meadows H, Duckworth MD, Kelsell RE, et al. Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res Mol Brain Res 2001;86:101-14.
- Meadows HJ, Benham CD, Cairns W, Gloger I, Jennings C, Medhurst AD, et al. Cloning, localisation and functional expression of the human orthologue of the TREK-1 potassium channel. Pflugers Arch 2000;439:714-22.
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honorè E. Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J Biol Chem 1999; 274: 26 691–6.
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honorè E. Lysophospholipids open the two-pore domain mechano-gated K channels TREK-1 and TRAAK. J Biol Chem 2000; 275: 10 128–33.
- Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, et al. TREK-1 is a heat-activated background K+ channel. EMBO J 2000;19:2483-91.
- Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thümmler S, Peng XD, et al. Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat Neurosci 2006;9:1134-41.
- Alloui A, Zimmermann K, Mamet J, Duprat F, Noël J, Chemin J, et al. TREK-1, a K+ channel involved in polymodal pain perception. EMBO J 2006;25:2368-76.
- Zhu YG, Chen XC, Chen ZZ, Zeng YQ, Shi GB, Su YH, et al. Curcumin protects mitochondria from oxidative damage and attenuates apoptosis in cortical neurons. Acta Pharmacol Sin 2004;25:1606-12.
- Wei T, Ni Y, Hou J, Chen C, Zhao B, Xin W. Hydrogen peroxide-induced oxidative damage and apoptosis in cerebellar granule cells: protection by Gingko biloba extract. Pharmacol Res 2000;41:427-33.
- Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma 2000;17:871-90.
- Espey MG, Miranda KM, Feelisch M, Fukuto J, Grisham MB, Vitek MP, et al. Mechanisms of cell death governed by the balance between nitrosative and oxidative stress. Ann NY Acad Sci 2000;899:209-21.
- Achike FI, Kwan CY. Nitric oxide, human diseases and the herbal products that affect the nitric oxide signalling pathway. Clin Exp Pharmacol Physiol 2003;30:605-15.
- Youdim MB, Lavie L, Riederer P. Oxygen free radicals and neurodegeneration in Parkinson’s disease: a role for nitric oxide. Ann N Y Acad Sci 1994;738:64-8.
- Fridovich I. Superoxide anion radical (O), superoxide dismutases, and related matters. J Biol Chem 1997; 272: 18 515–7.
- Hansen MB, Nielson SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods 1989;119:203-21.
- Yuan Q, Jiang DJ, Chen QQ, Wang S. Role of asymmetric dimethylarginine in homocysteine-induced apoptosis of vascular smooth muscle cells. Biochem Biophys Res Commun 2007;356:880-5.
- Jeong DW, Kim TS, Cho IT, Kim IY. Modification of glycolysis affects cell sensitivity to apoptosis induced by oxidative stress and mediated by mitochondria. Biochem Biophys Res Commun 2004;313:984-91.
- Konishi H, Matsuzaki H, Takaishi H, Yamamoto T, Fukunaga M, Ono Y, et al. Opposing effects of protein kinase C delta and protein kinase B alpha on H2O2-induced apoptosis in CHO cells. Biochem Biophys Res Commun 1999;264:840-6.
- Liniger R, Popovic R, Sullivan B, Gregory G, Bickler PE. Effects of neuroprotective cocktails on hippocampal neuron death in an in vitro model of cerebral ischemia. J Neurosurg Anesthesiol 2001;13:19-25.
- Marklund L, Henriksson R, Grankvist K. Cisplatin-induced apoptosis of mesothelioma cells is affected by potassium ion flux modulator amphotericin B and bumetanide. Int J Cancer 2001;93:577-83.
- Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci 2003;23:4798-802.
- Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, et al. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 1997;278:114-7.
- Wang L, Zhou P, Craig RW, Lu L. Protection from cell death by mcl-1 is mediated by membrane hyperpolarization induced by K+ channel activation. J Membr Biol 1999;172:113-20.
- Zhao YM, Sun LN, Zhou HY, Wang XL. Voltage-dependent potassium channels are involved in glutamate-induced apoptosis of rat hippocampal neurons. Neurosci Lett 2006;398:22-7.
- Lee AL, Dumas TC, Tarapore PE, Webster BR, Ho DY, Kaufer D, et al. Potassium channel gene therapy can prevent neuron death resulting from necrotic and apoptotic insults. J Neurochem 2003;86:1079-88.