
Synergistic effects of prostaglandin E1 and lithium in a rat model of cerebral ischemia1
Introduction
Development of neuroprotective agents against ischemia-induced brain damage has been a therapeutic strategy to reduce the mortality and morbidity associated with stroke[1]. Heat shock proteins (HSPs) are molecular chaperones that bind to unfolded or misfolded proteins to ensure proper folding and prevent intracellular protein aggregation[2]. It has been demonstrated that HSPs such as HSP70 and heme oxygenase-1 (HO-1) are important regulators of cellular survival and may be used as potential therapeutic targets against ischemic neuronal injury[3–5].
Prostaglandin E1 (PGE1) has several pharmacological effects, including cytoprotection, vasodilation, inhibition of platelet aggregation, membrane stabilization and anti-inflammation, etc[6]. The clinical uses of PGE1 are very broad and include treatment of ischemic diseases such as cerebral, myocardial and hepatic ischemia. In recent studies, Matsuo reported that PGE1 induced HSP70, glucose-regulated protein 78 (GRP 78) and HSP86 immediately after hepatic ischemia reperfusion. HSPs might therefore, play an important role in mediating protective actions of PGE1 against ischemia/reperfusion injury of the liver[7]. We speculate that PGE1 may also induce HSPs and protect neurons against ischemic damage in rodent models of cerebral ischemia.
Lithium has been extensively used in the treatment of bipolar mood disorder[8]. Growing evidence suggests that lithium is a neuroprotective drug that is effective against a variety of insults, such as glutamate-induced excitotoxicity, ischemia-induced neuronal damage and other neurodegenerative conditions[9]. Recently, Ren et al[10] reported that treatment of rats with lithium decreased the infarct volume in a permanent focal cerebral ischemia model and the neuroprotective effects of lithium were associated with upregulation of cytoprotective HSP70 in the ischemic brain hemispheres. Moreover, research from our laboratory[11,12] also found that lithium could potentiate the neuroprotective effects of PGA1 (another prostanoid bioactive compound) through upregulation of HSPs.
As both PGE1 and lithium are clinical therapeutic drugs, the present study was undertaken to explore whether lithium could enhance the neuroprotection of PGE1 in a rat ischemic model with clinically achievable methods of drug administration and whether the synergetic effects of lithium and PGE1 take place at the level of HSPs.
Materials and methods
Animal preparation and experimental protocol Male Sprague–Dawley rats weighing 280–300g were purchased from the Center for Experimental Animals, Soochow University. The NIH guidelines for Care and Use of Laboratory Animals were followed in all animal procedures. In the drug treatment stud, three batches of rats were used. The rats in each batch were randomly divided into 7 groups: sham-operated group, permanent middle cerebral artery occlusion (pMCAO) group, lithium+pMCAO, PGE1 8 μg/kg+pMCAO, PGE1 16 μg/kg+pMCAO, PGE1 8 μg/kg+lithium+pMCAO, and PGE1 16 μg/kg+lithium+pMCAO groups. The first batch of 70 rats was used for evaluation of infarct volume, brain water content and neurological deficits. The second batch of 70 rats was used for monitoring regional cerebrocortical blood flow (rCBF) and immunoblotting, and the last batch of 70 rats was used for detecting body temperature and immunohistochemistry. In addition, 50 rats were used to study the time-course of HSPs protein expression, these rats were killed 1, 3, 6, 12, and 24 h after pMCAO onset and the HSPs protein expression in the ipsilateral striatum was detected by immunoblotting.
Rat pMCAO model and treatment with lithium and PGE1 The rat pMCAO model was produced using the intraluminal suture technique as described by Longa with minor modifications[13,14]. Rats were anesthetized by 4% chloral hydrate (350 mg/kg). A 30 mm length of monofilament nylon suture (Φ0.22–0.24 mm) with rounded tip was inserted to the internal carotid artery through a small incision in the right common carotid artery and then advanced to the Circle of Willis. The suture remained there until the rats were killed. Body temperature was closely monitored with a rectal probe and maintained in the range of 37.0±0.5 °C with a heating pad during and after surgery until recovery from anesthesia. Sham-operated rats underwent the same procedures except for the pMCAO. About 20%–30% of rats died 24 h after ischemia onset and were excluded from further analysis. Those rats showing tremor and seizure were also excluded from further analysis, with an incidence less than 5%. For all experiments, at indicated doses, lithium was given subcutaneously to rats twice (the first dose was given 24 h before and the second dose was given immediately before the onset of pMCAO, and PGE1 (dissolved in normal saline) was injected intravenously to rats once immediately after the pMCAO. Sham-operated animals received injections of normal saline. Body temperature was measured 5 min, 2 h and 24 h after pMCAO onset with a rectal probe.
Detection of regional cerebrocortical blood flow Laser-Doppler flowmetry (LDF, ML191 Laser Doppler Blood FlowMeter, Sydney, Australia) was used to monitor rCBF. After the rats were placed in a stereotactic frame, a hole of 1 mm diameter was drilled on the right side, 5 mm lateral and 1.5 mm posterior to the Bregma, and the LDF probe was placed into the burr hole (region of blood supply by cerebral middle brain artery). The rCBF was detected prior to onset of ischemia to acquire the pre-ischemia blood flow level. Five minutes after the pMCAO operation, the probe was inserted again to monitor rCBF, showing a sharp drop of rCBF (approximately 5%–10% of the pre-ischemia value). Then the rCBF was measured again 15, 30 and 60 min after surgical operation[15].
Evaluation of infarct volume, brain water content and neurological deficit Twenty-four hours after ischemia, the neurological deficits in rats subjected to pMCAO were evaluated using a protocol previously described in a blinded manner[16]. The total score of 10 was evaluated as follows: (1) When rats were suspended by the tail, the left forelimb was flexed, scored 1–4 according to the severity; (2) When rats were placed on a smooth plane, the lateral push resistance toward the left side decreased, scored 1–3. (3) The rats were pulled gently backward by the tail, the left forelimb showed decreased strength, scored 1–3. After being scored, rats were killed and the brains were dissected and sliced in a plastic module (Harvard Apparatus, MA, USA 3-mm thickness) and stained with 4% 2,3,5-triphenyltetrazolium chloride (TTC) for 30 min and then fixed with 4% paraformaldehyde. The total wet weight of the TTC stained brains was quantified with an electronic scale (Mettler-Toledo Group, OH, USA). The wet red and white brain regions of the TTC-stained brains were collected separately. Infarct volume was analyzed using 5 slices of 3-mm coronal sections from each brain and calculated with the following formula: infarct volume=(total wet weight–red weight)/ total wet weight×100%. After the wet weight of the brains was quantified, the red and white parts of these brains were desiccated at 105 °C for 48 h until the weight was constant. The total weight of the dried TTC-stained brains was obtained, and the water content of each brain was measured as follows[17]: water content=(wet weight–dried weight)/wet weight×100%.
Immunoblotting Immunoblotting was carried out as previously described[18]. Brain tissues from the ischemic striatum of the right middle cerebral artery territory and the corresponding area of sham-operated rats were homogenized and proteins were extracted with a lysis buffer [10 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 1% Triton-100, 0.1% SDS, 5 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 0.28 U/mL aprotinin, 50 µg/mL leupeptin, 1 mmol/L benzamidine, 7 µg/mL pepstatin A]. Protein concentrations were determined (SmartSpec3000 Spectrophotometer, Bio-Rad, Hercules, CA, USA) using a BCA kit (Pierce, Rockford, IL, USA). A 50 µg aliquot of protein from each sample was separated using 10% SDS-PAGE and subsequently transferred to a nitrocellulose membrane. Afterwards, the membranes were incubated with the specific antibodies against HSP70 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and HO-1 (Santa Cruz Biotechnology, 1:200) at 4 °C for 3 h, then incubated with a horseradish peroxidase-conjugated secondary antibody (1:5000; Sigma, St Louis, MO, USA) at room temperature for 1 h. Immunoreactivity was detected by an enhanced chemiluminescent autoradiography (ECL kit; Amersham, Piscataway, NJ, USA) in accordance with the manufacturer’s instructions. The membranes were reprobed with β-actin (1:5000; Sigma) after striping with Tris-buffered saline containing 0.1% Tween-20 (TBST) and 2% β-mecaptoethanol, 65 °C, 1 h.
Immunohistochemistry Immunohistochemistry was carried out as previously described with minor modifications[11]. Coronal sections of 20 μm thickness were cut with a cryostat, fixed with absolute ethanol for 15 min, incubated with phosphate-buffered saline (PBS, pH 7.4) containing 0.1% Triton X-100 and 1% bovine serum albumin (BSA) for 1 h, and then rinsed with PBS 3 times. Brain sections were then incubated with a mouse monoclonal immunoglobulin G (IgG) against HSP70 (1:200; Santa Cruz Biotechnology) in a humidified container at 4 °C overnight. The sections were rinsed 3 times in PBS and sequentially incubated with fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (1:400, Santa Cruz Biotechnology) in a humidified container at 4 °C for 1 h. Sections were then washed 3 times in PBS and incubated with 0.5 μg/mL 4,6-diamidino-2-phenylindole (DAPI) at 4 °C for 10 min. Sections were then washed in PBS and sealed with a coverslip. The slides were analyzed with a laser confocal microscope (Nikon D-Eclipse C1, Tokyo, Japan).
Statistics analysis Statistical analysis was carried out by one-way ANOVA. The intergroup comparisons (post-hoc analysis) among the data with equal variances were carried out with the least significant difference (LSD) method, while Tamhane’s T2 method was used for the data with unequal variances. P<0.05 was considered to be significant.
Results
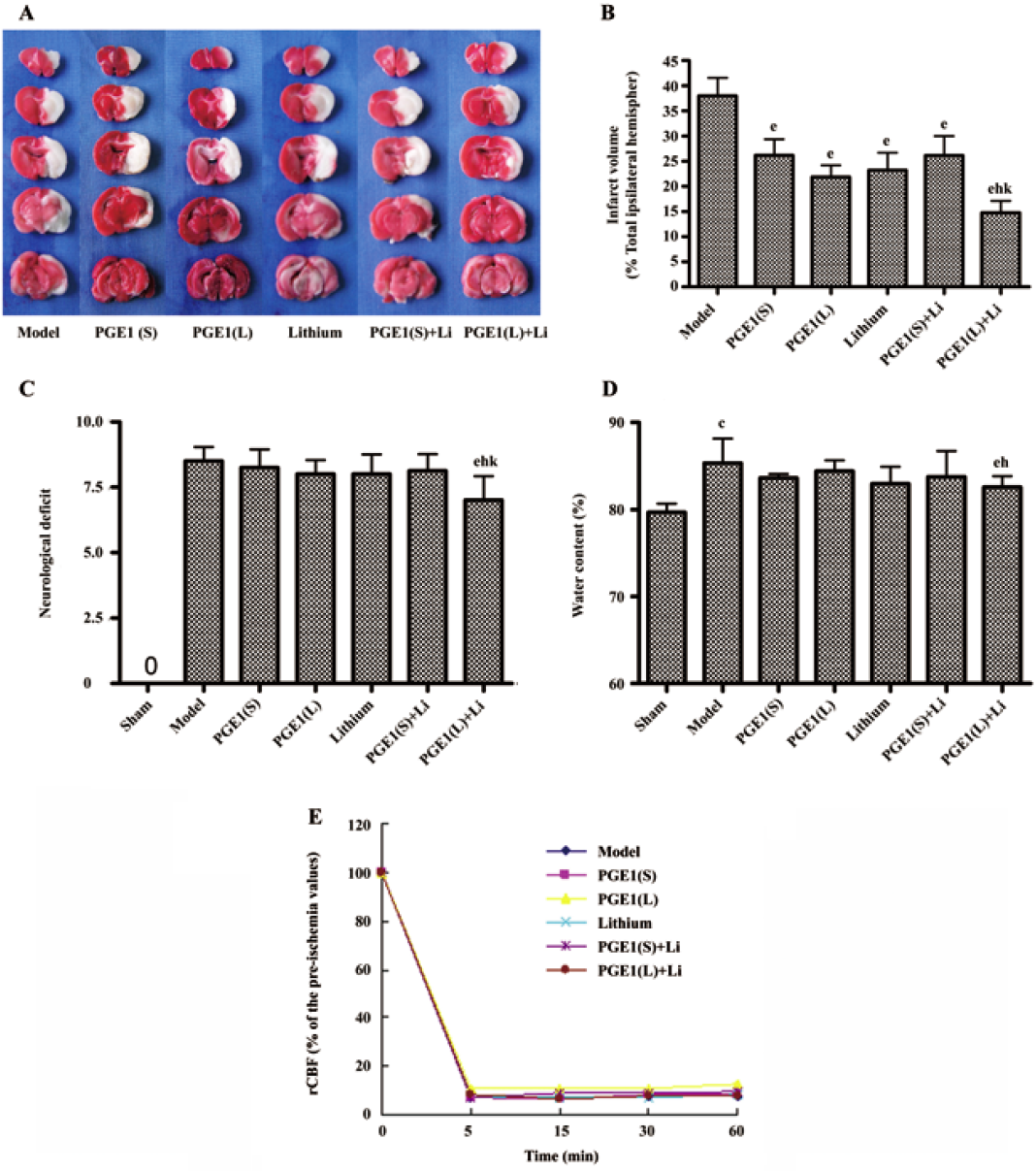
Effects of PGE1 and lithium on rCBF Five minutes after the pMCAO operation, in all of the rats subjected to pMCAO, the rCBF decreased to approximately 5%–10% of the pre-ischemia values and remained low thereafter. PGE1, lithium and their combination had no statistically significant improvement on reduction in rCBF 5 min, 15 min, 30 min and 60 min after the surgical operation (Figure 1E).
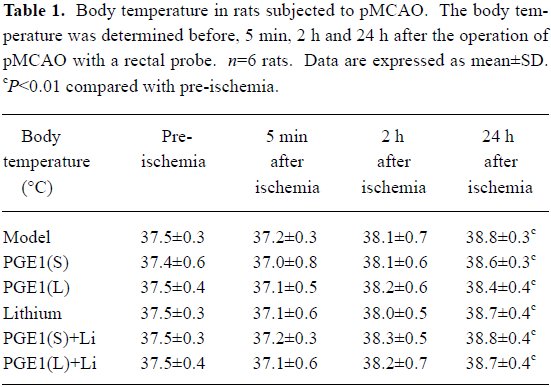
Effects of PGE1 and lithium on body temperature The body temperature of rats showed no significant difference 5 min and 2 h after the pMCAO operation, whereas the body temperature increased significantly 24 h after pMCAO (P<0.01 vs pre-ischemia, Table 1). However, there is no significance between the model group and drug treatment groups at all of these time points after pMCAO.
Full table
PGE1 and lithium reduced pMCAO-induced brain damage In rats subjected to pMCAO for 24 h, extensive infarction was detected in the cerebral cortical and subcortical areas over a series of brain sections. Treatment with lithium (0.5 mEq/kg, sc) starting 2 days before the onset of pMCAO, or a single intravenous injection of PGE1 (8 and 16 µg/kg) immediately after the onset of pMCAO resulted in a significant reduction in infarct volume as detected by TTC staining (P<0.05 vs model group). Furthermore, co-administration of lithium (0.5 mEq/kg, sc) and PGE1 (16 µg/kg, iv) produced a greater reduction in infarct volume (P<0.05 vs PGE1 16 µg/kg) (Figure 1A and 1B). In addition, the water content in rat brains subjected to pMCAO was significantly increased, and these rats showed robust motor behavioral deficits. Although administration of lithium (0.5 mEq/kg) alone or PGE1 (8 and 16 µg/kg) alone had no significant effect on brain water content and behavioral deficits, combination of PGE1 16 µg/kg and lithium 0.5 mEq/kg significantly decreased ischemia-induced increase in brain water content and neurological deficits (P<0.05 vs model group, P<0.05 vs PGE1 16 µg/kg) (Figure 1C, 1D).
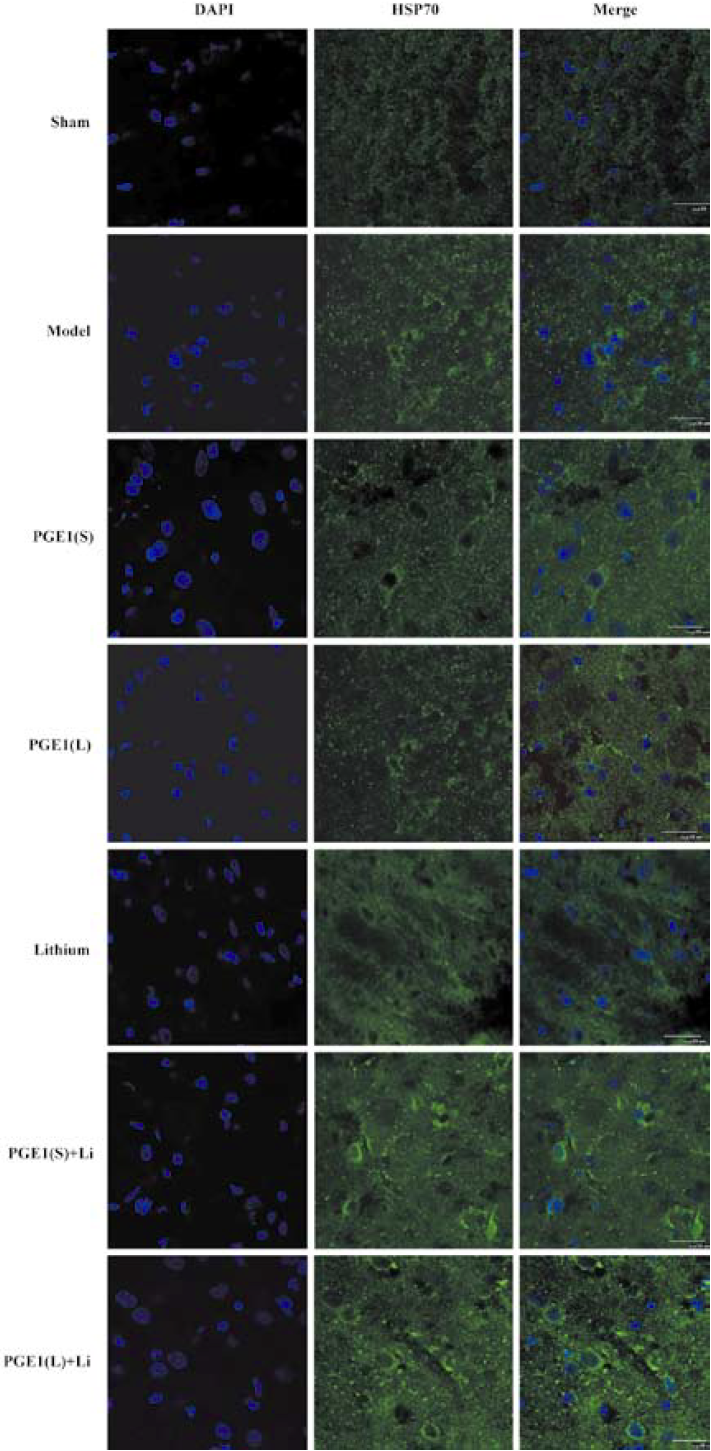
Enhanced induction of HSP70 by PGE1 and lithium In immunoblotting, the expression of HSP70 was significantly upregulated 24 h after the onset of ischemia in the ischemic striatum (Figure 2A). PGE1 (8, 16 µg/kg) or lithium (0.5 mEq/kg) alone showed no significant effect on HSP70 levels. However, a combination of PGE1 and lithium significantly increased HSP70 protein levels compared with both the model group and the PGE1 8 µg/kg group (P<0.05, Figure 2B). There were few HSP70-positive cells in the striatum of sham-operated rats as detected by immunohistochemistry. Increased numbers of HSP70-positive cells were observed 24 h after pMCAO. However, a combination of PGE1 and lithium further increased the number of HSP70-positive cells. Those cells with high levels of HSP70 appeared to have relatively normal morphology Figure 3.
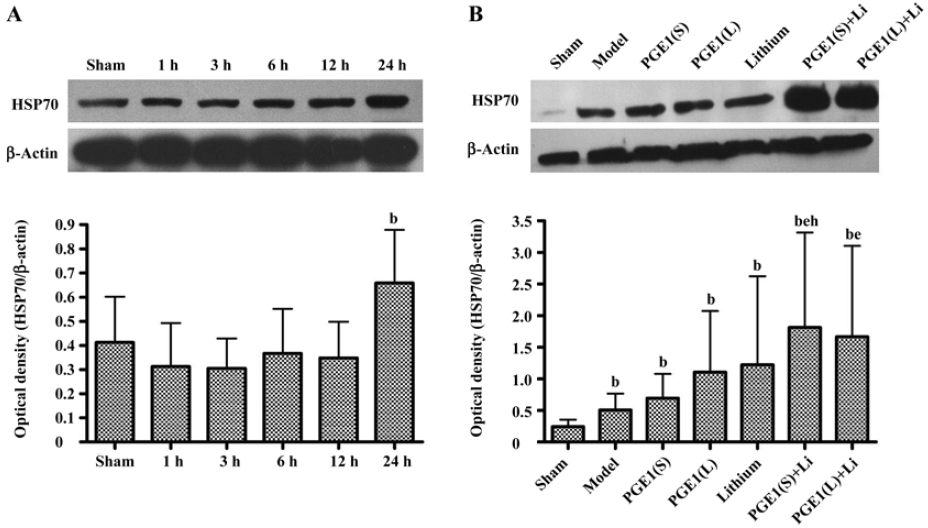
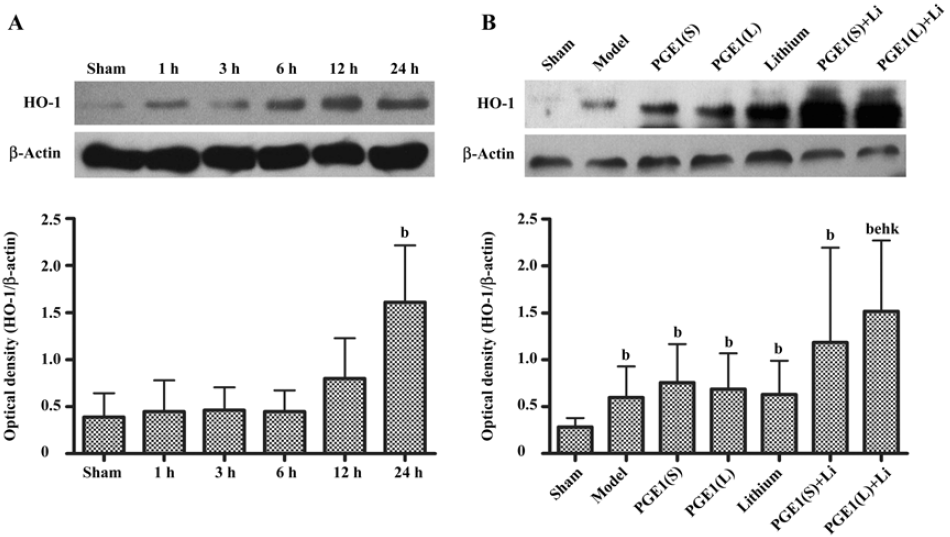
Enhanced induction of HO-1 by PGE1 and lithium The expression of HO-1 was significantly upregulated 24 h after the onset of ischemia in the ischemic striatum (Figure 4A). PGE1 (8, 16 µg/kg) or lithium (0.5 mEq/kg) alone showed no significant effect on HO-1 protein levels. However, a combination of lithium and PGE1 16 µg/kg elicited a significant increase in HO-1 levels compared with both the model group and the PGE1 16 µg/kg alone group (P<0.05, Figure 4B).
Discussion
After pMCAO, the rats showed significant motor behavioral deficits; extensive infarction was detected in the cerebral cortical and subcortical regions and water content in the brain was significantly increased. In the groups treated with PGE1 or lithium alone, reduction in infarct volume was observed. A greater reduction in infarct volume was seen in rats given combined PGE1 and lithium. Moreover, the benefits were extended to brain edema and neurological deficits with the combination of lithium and PGE1. These results suggest that lithium can enhance the neuroprotective effects of PGE1.
Previous studies reported that lithium had no effect on cerebrocortical blood flow in animal models of ischemia[10]. Although PGE1 has vessel dilation effects[6,19], when we monitored cerebrocortical blood flow after drug treatment, neither PGE1 alone nor combination of PGE1 with lithium significantly influenced the cerebrocortical blood flow of the pMCAO animals. This difference may be due, in part, to lower PGE1 doses and different administration routes in these experiments[18]. Thus, neuroprotection exerted by PGE1 and lithium in the rat brain ischemia model in this study could not be the result of increased cerebrocortical blood flow. Temperature is another critical variable in intraluminal filament occlusion models in which hyperthermia is frequently observed due to hypothalamic ischemia[14]. Therefore, drug effects on temperature regulation were observed 5 min, 2 h and 24 h after pMCAO onset in the study. The results showed that the body temperature showed no significant difference 5 min and 2 h after the pMCAO operation, whereas the body temperature increased significantly 24 h after pMCAO. However, there is no significant difference between model group and drug treatment groups at all of these time points after pMCAO. These results suggest that PGE1 and lithium exert their neuroprotection on cerebral ischemia independent of body temperature.
Heat shock proteins are molecular chaperones that regulate folding of nascent and denatured proteins, transport proteins through subcellular compartments and modify the activity of proteins by altering conformational state[2]. A number of studies have documented that levels of HSPs are increased in the ischemic penumbra of brain in animal models of focal ischemia, where many injured neurons survive[20]. HSPs may exert their neuroprotective effects by antagonizing apoptosis-inducing factor[21]. Induction of HSPs also plays a role in preconditioning-induced resistance of neurons to ischemic insults[22].
HSP70 and HO-1 are important members of the heat shock protein family[3–5]. It is reported that gene transfer induced HSP70 overexpression protects neurons from ischemic brain damage in experimental rat stroke models[23]. Moreover, overexpression of HSP70 inhibits the activation of NF-κB, which is persistently activated during ischemia and appears to promote apoptotic cell death[24]. On the contrary, the deletion of the HSP70 gene remarkably increases cytochrome c release into the cytoplasm and subsequent caspase-3 activation, thereby exacerbating apoptosis and increasing infarction volume after focal cerebral ischemia[25]. HO-1 is the enzyme that converts the heme molecule into bilirubin, carbon monoxide, and iron. It serves as an adaptive mechanism to protect the brain from ischemic injury as evidenced by studies conducted on transgenic mice that overexpress HO-1 in neurons[26]. In our ischemic model, HSP70 and HO-1 protein levels were maximally increased at 24 h post-insult in the ischemic striatum. Although Ren et al[10] reported that neuroprotective effects of lithium (1 mEq/kg) were associated with upregulation of cytoprotective HSP70 in the ischemic brain hemispheres, and Matsuo found that PGE1 (3 µg/kg) induced HSP70 and HSP86 after hepatic ischemia reperfusion in mice by both microarray analysis and real time-PCR, we found no significant increase in HSPs when treated with PGE1 8–16 μg/kg or lithium 0.5 mEq/kg alone. This difference may be due, in part, to lower dose of lithium, different analytical methods and different rodent models used. However, co-administration with PGE1 and lithium robustly enhanced the increase in HSP70 and HO-1 protein levels in the ischemic striatum. Therefore, induction of HSPs by lithium and PGE1 together may play an important role in protecting against ischemia-induced neuronal injury.
It is possible that other molecular and cellular actions may also participate in PGE1 and lithium-induced neuroprotection in the pMCAO model. These include the effects of PGE1 on cytoprotection, inhibiting platelet aggregation, membrane stabilization and anti-inflammation, etc[6,18]; and lithium’s ability to inhibit N-methyl-D-aspartate receptors[27], upregulate cytoprotective Bcl-2 [28], and induce the expression of brain-derived neurotrophic factor in discrete brain areas[29].
In previous work, research from our laboratory[11,12] indicated that lithium could potentiate the neuroprotective effects of PGA1 through upregulation of HSPs. However, PGA1 can only be given through intracerebral ventricle administration, which is impossible for use in clinics. Therefore, we studied PGE1, a homolog of PGA1, which can be given conveniently through intravenous injection. The similar synergistic neuroprotection on cerebral ischemia was also found by combination therapy with PGE1 and lithium. As PGE1 and lithium are both used with clinically achievable methods of drug administration in this study, the combination therapy of PGE1 and lithium has more practical application value than PGA1 and lithium, which might support a potential clinical therapy for ischemic cerebrovascular diseases.
In conclusion, the present study shows that although some neuroprotective effects were obtained with adminis-tration of PGE1 or lithium alone, their combination ex-hibited greater neuroprotection. Combined PGE1 and lithium treatment significantly enhanced the expression of HSP70 and HO-1 in the ischemic striatum. As PGE1 and lithium are drugs used in clinics, these results strongly support a potential use in clinical therapy for ischemic cerebrovascular diseases.
References
- Onteniente B, Rasika S, Benchoua A, Guegan C. Molecular pathways in cerebral ischemia: cues to novel therapeutic strategies. Mol Neurobiol 2003;27:33-72.
- Ohtsuka1 K, Suzuki T. Roles of molecular chaperones in the nervous system. Brain Res Bull 2000;53:141-6.
- Tsuchiya D, Hong S, Matsumori Y, Shiina H, Kayama T, Swanson RA, et al. Overerexpression of rat heat shock protein 70 is associated with reduction of early mitochondrial cytochrome C release and subsequent DNA fragmentation after permanent focal ischemia. J Cereb Blood Metab 2003;23:718-27.
- Majda BT, Meloni BP, Rixon N, Knuckey NW. Suppression subtraction hybridization and northern analysis reveal upregulation of heat shock, trkB, and sodium calcium exchanger genes following global cerebral ischemia in the rat. Mol Brain Res 2001;93:173-9.
- Geddes JW, Pettigrew LC, Holtz ML, Craddock SD, Maines MD. Permanent focal and transient global cerebral ischemia increase glial and neuronal expression of heme oxygenase-1, but not heme oxygenase-2, protein in rat brain. Neurosci Let 1996;210:205-8.
- Schror K, Hohlfeld T. Mechanisms of anti-ischemic action of prostaglandin E1 in peripheral arterial occlusive disease. Vasa 2004;33:119-24.
- Matsuo K, Togo S, Sekido H, Morita T, Kamiyama M, Morioka D, et al. Pharmacologic preconditioning effects: prostaglandin E1 induces heat-shock proteins immediately after ischemia/reperfusion of the mouse liver. J Gastrointest Surg 2005;9:758-68.
- Bauer M, Alda M, Priller J, Young LT. International Group For The Study Of lithium Treated Patients (IGSLI). Implications of the neuroprotective effects of lithium for the treatment of bipolar and neurodegerative disorders. Pharmacopsychiatry 2003;36:S250-4.
- Chuang DM, Chen RW, Chalecka-Franaszek E, Ren M, Hashimoto R, Senatorov V, et al. Neuroprotective effects of lithium in cultured cells and animal models of diseases. Bipolar Disord 2002;4:129-36.
- Ren M, Senatotov VV, Chen RW, Chuang DM. Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc Natl Acad Sci USA 2003;100:6210-5.
- Xu XH, Zhang HL, Han R, Gu ZL, Qin ZH. Enhancement of neuroprotection and heat shock protein induction by combined prostaglandin A1 and lithium in rodent models of focal ischemia. Brain Res 2006;1102:154-62.
- Xu XH, Hua YN, Zhang HL, Wu JC, Miao YZ, Han R, et al. Greater stress protein expression enhanced by combined prostaglandin A1 and lithium in a rat model of focal ischemia. Acta Pharmacol Sin 2007;28:1097-104.
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989;20:84-91.
- Li F, Omae T, Fisher M. Spontaneous hyperthermia and its mechanism in the intraluminal suture middle cerebral artery occlusion model of rats. Stroke 1999;30:2464-71.
- Zhang Y, Wang L, Li J, Wang XL. 2-(1-Hydroxypentyl)-benzoate increases cerebral blood flow and reduces infarct volume in rats model of transient focal cerebral ischemia. J Pharmacol Exp Ther 2006;317:973-9.
- Tamura A, Graham DI, McCulloch J, Teasdale GM. Focal cerebral ischemia in the rat. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J Cereb Blood Flow Metabol 1981;1:53-60.
- Rosenberg GA, Scremin O, Estrada E, Kyner WT. Arginine vasopressin V1-antagonist and atrial natriuretic peptide reduce hemorrhagic brain edema in rats. Stroke 1992;23:1767-73.
- Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, Chase TN. Nuclear factor kappar B nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor-mediated apoptosis in rat stratium. J Neurosci 1999;19:4023-33.
- Abe H, Sugino N, Matsuda T, Ueda Y, Mori H, Aono M. Effects of prostaglandin E1 on transient forebrain ischemia, especially in hippocampal CA 1 regions of the gerbil. Masui 1996;45:1216-22.
- Wagstaff MJD, Collaco-Moraes Y, Aspey BS, Coffin RS, Harrison MJG, Latchman DS, et al. Focal cerebral ischaemia increases the levels of several classes of heat shock proteins and their corresponding mRNAs. Mol Brain Res 1996;42:236-44.
- Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol 2001;3:839-43.
- Kim H, Huh PW, Kim C, Kim YJ, Park EM, Par YM. Cerebral activation and distribution of inducible hsp110 and hsp70 mRNAs following focal ischemia in rat. Toxicology 2001;167:135-44.
- Li CY, Lee JS, Ko YG, Kim JI, Seo JS. Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and up stream of caspase-3 activation. J Biol Chem 2000; 275: 25 665 – 71.
- Feinstein DL, Galea E, Aquino DA, Li GC, Xu H, Reis DJ. Heat shock protein 70 suppresses astroglial-inducible nitric-oxide synthase expression by decreasing NFkappaB activation. J Biol Chem 1996; 271: 17 724–32.
- Lee SH, Kwon HM, Kim YJ, Lee KM, Kim M, Yoon BW. Effects of HSP70.1 gene knockout on the mitochondrial apoptotic pathway after focal cerebral ischemia. Stroke 2004;35:2195-9.
- Panahian N, Yoshiura M, Maines MD. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem 1999;72:1187-203.
- Ma J, Zhang GY. Lithium reduced N-methyl-D-aspartate receptor subunit 2A tyrosine phosphorylation and its interactions with Src and Fyn mediated by PSD-95 in rat hippocampus following cerebral ischemia. Neurosci Lett 2003;348:185-9.
- Chen G, Zeng WZ, Yuang PX, Huang LD, Jiang YM, Zhao ZH. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J Neurochem 1999;72:879-82.
- Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the brain. Psychopharmacology 2001;158:100-6.