
Felodipine attenuates vascular inflammation in a fructose-induced rat model of metabolic syndrome via the inhibition of NF-κB activation1
Introduction
The metabolic syndrome consists of the combined presentation of multiple cardiovascular risk factors that include abdominal obesity, hypertension, insulin resistance, hyperinsulinemia, and dyslipidemia[1,2]. Although the definition and nomenclature of metabolic syndrome is controversial, this syndrome is now widely recognized as a distinct pathological entity[3]. Recently-published data have suggested that patients with metabolic syndrome are at higher risk of cardiovascular morbidity and mortality[4].
Clinical studies have shown that patients with metabolic syndrome are more prone to atherosclerosis than normal patients[5], even in young adults[6]. Recently, Delbosc et al reported that the mesenteric arterial media/lumen ratio was higher in fructose-fed rats (FFR)[7]. Those interesting findings suggested that structural changes in the vascular wall may be involved in the cardiovascular alterations associated with metabolic syndrome.
It is well established that inflammation plays a pivotal role in the development of classical atherosclerosis, and the inhibition of the expression of adhesion molecules reduces the plaque volume in animal models, probably by reducing the recruitment of monocytes from the blood[8,9]. Patients with primary hyperglyceridemia display increased plasma levels of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1)[10]. Moreover, hyperglycemia induces the increased expression of adhesion molecules in cultured endothelial cells[11]. Thus it is tempting to speculate that the propensity to accelerated lesion formation in metabolic syndrome may involve increased vascular inflammatory response. However, it is not well known whether metabolic syndrome affects the expression of adhesion molecules in the arterial wall before the lesion formation.
L-type calcium channel blockers (CCB) are widely used to treat patients with hypertension and atherosclerosis[12,13]. In clinical trials, CCB have been proven to reduce the rate of cardiovascular events and inhibit the progression of atherosclerosis[12,13]. It is suggested that CCB can be used to treat patients with metabolic syndrome[3]. However, few studies have focused on the effects of CCB on vascular changes in metabolic syndrome.
Therefore, we hypothesized that the infiltration of macrophages and the expression of adhesion molecules are increased, and felodipine, a member of L-type CCB, may inhibit vascular inflammation in the aorta of high FFR. To test our hypothesis, high FFR were used to demonstrate hypertension, insulin resistance, and hypertriglyceridemia[14,15].
Material and methods
Reagents Felodipine was provided by AstraZeneca Pharmaceutical (Shanghai, China). The polyclonal antibodies against rat ICAM-1 (sc-1511) and rat VCAM-1 (sc-8304) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The monoclonal antibody against rat CD68 was purchased from Lab Vision (Fremont, CA, USA). The quantitative real-time RT-PCR system was obtained from TaKaRa Biotechnology (Dalian, China).
Animals and experimental design All animal care and experimental protocols complied with the guidelines of the National Institutes of Health and the guidelines for the care and use of laboratory animals of Qi-Lu hospital (Ji-nan, China). Male Wistar rats (6 weeks old) were obtained from the Center for Experimental Animals, Shandong University School of Medicine (Grade II Certificate No 0004170; Ji-nan, China). The rats were fed a standard commercial chow diet ad libitum and housed in a room under controlled temperature (23–25 °C), humidity, and a 14 h light/10 h dark cycle throughout the entire experiment. The animals were randomly divided into 2 groups. The control rats (n=12) were fed normal chow and given normal water, and the FFR (n=18) were fed normal chow and given water containing 10% (w/v) fructose[14–17]. Using water containing 10% fructose to feed animals has been proven to be an effective and stable method to induce metabolic syndrome in rats[14] and hamsters[18].
After being on the diet for 32 weeks, the FFR were further divided into 2 groups: the FFR group (n=9) and the FFR+F group (n=9). The FFR+F group rats received 5 mg·kg-1·d-1 felodipine by gavage for 6 weeks. The rats of the control and FFR groups received an equal volume of vehicle by gavage. The selection of felodipine dosage was based on a previous study that demonstrated that felodipine reduces blood pressure effectively with the given dosage in spontaneous hypertensive rats[19].
Body weight and tail blood pressure were measured once per week. Blood samples were collected from the jugular sinus at the beginning of the study, after 32 weeks of the fructose diet, and at the end of study from all the rats for relative analysis. At the end of 38 weeks, the rats were killed by a pentobarbital overdose, and the aorta was aseptically excised for measurement.
Systolic blood pressure measurement Systolic blood pressure (SBP) was estimated by a tail-cuff method in conscious animals as previously described[15,20]. The reported blood pressure value was the mean of 5 systolic measurements.
Biochemical measurements Serum glucose, total cholesterol, and triglycerides were measured using an enzymatic method with an auto-analyzer (ADVIA 1650; Bayer, Germany). Serum insulin was determined by a radio-immunoasssy kit (Dongya, Beijing, China). Insulin resistance was calculated by the homeostasis model assessment (HOMA) method[15].
Immunohistochemistry The upper part of the descending aorta (approximate 0.5 cm) was fixed in 4% formaldehyde, embedded in paraffin, and cut into 5 μm-thick sections for immunohistochemical staining using standard techniques. In brief, endogenous peroxidase activity was inhibited by incubation with 3% H2O2. Sections were blocked with 5% bovine serum albumin in phosphate-buffered saline (PBS) and incubated overnight at 4 °C with primary antibodies. The primary antibodies included rabbit antirat ICAM-1 diluted at 1:150 and rabbit anti-rat VCAM-1 diluted at 1:200 to detect ICAM-1 and VCAM-1, respectively. After washing with PBS, the sections were incubated with a secondary antibody at room temperature for 60 min. Visualization of a positive reaction was developed with use of a peroxidase substrate solution containing 0.02% H2O2 and 0.1% 3,3’-diaminobenzidine tetrahydrochloride (ZSBIO, Beijing, China) in the PBS to display a brown color in the reaction product. The sections were then counterstained with hematoxylin. Incubation with PBS instead of the primary antibody was used as a negative control.
To identify macrophages in the aorta, sections were blocked with 5% bovine serum albumen in PBS and incubated with a mouse antirat CD68 monoclonal antibody diluted at 1:150 for 2 h at room temperature. After washing with PBS, the slides were incubated with a fluorescein isothiocyanate-conjugated streptavidin secondary antibody at a dilution of 1:500 at room temperature for 60 min and rewashed. Sections were mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA) with DAPI counterstaining. Sections incubated with PBS without primary antibody served as controls.
Histopathological slides were analyzed by use of a computer-assisted morphometric analysis system (Image-Pro Plus 5.0; Media Cybernetics, Silver Spring, USA). Four areas from each arterial section were analyzed. The resulting values were used to obtain an average staining for each artery.
Quantitative real-time RT-PCR Total RNA was isolated from each rat aorta with TRIzol reagent (Invitrogen, USA). Total RNA was quantified by spectrophotometry and reverse transcribed with the use of the M-MLV reverse transcriptase system (Promega, Madison, WI, USA) and oligo(dT) (T 12–18) primers. Quantitative real-time RT-PCR was performed with the use of a LightCycler (Roche Applied Science, USA) involving the SYBR-based method in 20 μL reaction volume following the manufacturer’s instructions. The PCR primers for ICAM-1 were 5'-TTTCGATCTTCCGACTAGGG-3' (forward) and 5'-AGCTTCAGAGGCAGGAAACA-3' (reverse); for VCAM-1 they were 5'-GCGAAGGAAACTGGAGAAGACA-3' (forward) and 5'-ACACATTAGGGACCGTGCAGTT-3' (reverse); and for GAPDH they were 5'-TGCCAAGTATGATGACATCAAGAAG-3' (forward) and 5'-AGCCCAGGATGCCCTTTAGT-3' (reverse). Quantitative values were obtained from the threshold cycle value (Ct), the point at which a significant increase of fluorescence is first detected. The relative changes in gene expression were analyzed with the 2(-Delta Delta Ct) method[21]. The specificity of RT-PCR was checked by analyzing melting curves and by gel electrophoresis of the amplicons.
Electrophoretic mobility shift assay Nuclear extracts were prepared from the aortic tissue using a nuclear extraction kit according to the manufacturer’s instruction (Active Motif, USA). Protein quantification was done using a RC DC protein assay kit (Bio-Rad, Hercules, CA, USA), and electrophoretic mobility shift assay (EMSA) was carried out using the digoxigenin (DIG) gel shift kit (Roche, USA). Briefly, the NF-κB consensus oligonucleotide (5'-AGTTGAGGGGACT-TTCCCAGGC-3') was end-labeled with DIG. Nuclear extracts (10 μg) were incubated with either a DIG-labeled NF-κB probe or a DIG-labeled NF-κB probe plus a NF-κB unlabeled probe for competitive experiments in a buffer containing 1 μg poly d(IC), poly-L-lysine, 20 mmol/L HEPES (pH 7.6), 1 mmol/L EDTA,10 mmol/L (NH4)2SO4, 1 mmol/L dithiothreitol, and 30 mmol/L KCl (total volume, 20 mL). The samples were loaded on a 6% native polyacrylamide gel in Tris-borate-EDTA buffer. After transfer to nylon membranes and incubation with a anti-DIG antibody conjugated with alkaline phosphatase, gel mobility shift was visualized by incubation with a CSPD chemiluminescence reagent and exposure to X-ray film.
Statistical analysis Values are expressed as mean±standard deviation. One-sample Kolmogorov–Smirnov test was used to analyze the distribution of all quantitative variables. All variables were normally distributed and had equal variance. Student’s two-tailed t-test and ANOVA were used where appropriate. Pearson’s correlation coefficient was used to assess the correlation between the expression of the adhesion molecular and metabolic biomarkers. All statistical analyses were performed using SPSS 13.0 software (SPSS, Chicago, IL, USA), and the differences were considered significant at P<0.05.
Results
Experimental model The metabolic syndrome model was achieved by the administration of 10% fructose in drinking water to male Wistar rats during the 32 weeks. The FFR group showed increased levels of body weight, blood pressure, triglycerides, and insulin when compared with the control rats (Table 1). At 16 weeks, a slight increase in body weight was observed in the FFR group (552±71 g vs 493±54 g, P=0.026) than in the control group, and at 32 weeks, body weight was significantly higher (P=0.001). Blood pressure was increased in FFR after 2 weeks of fructose ingestion, and was further increased after 32 weeks of fructose ingestion (P<0.01). The FFR also showed a significant increase in triglycerides, insulin, and HOMA after 32 weeks, (P<0.01). No difference was observed in fasting glucose and total cholesterol between the 2 groups (Table 1).
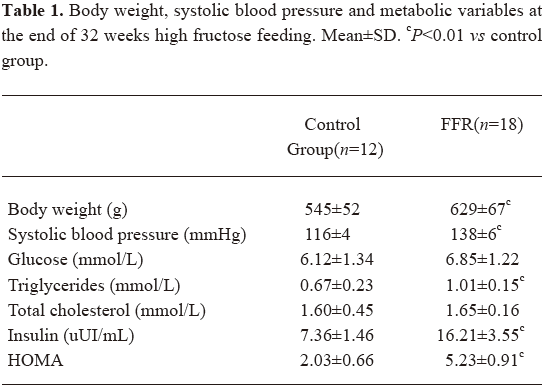
Full table
Effect of felodipine on the metabolic parameters in FFR After 6 weeks of treatment with felodipine, blood pressure was reduced significantly. Insulin and HOMA were markedly reduced in the felodipine group (P<0.05 and P<0.01). No difference was observed in serum lipids and glucose when compared with FFR (Table 2).
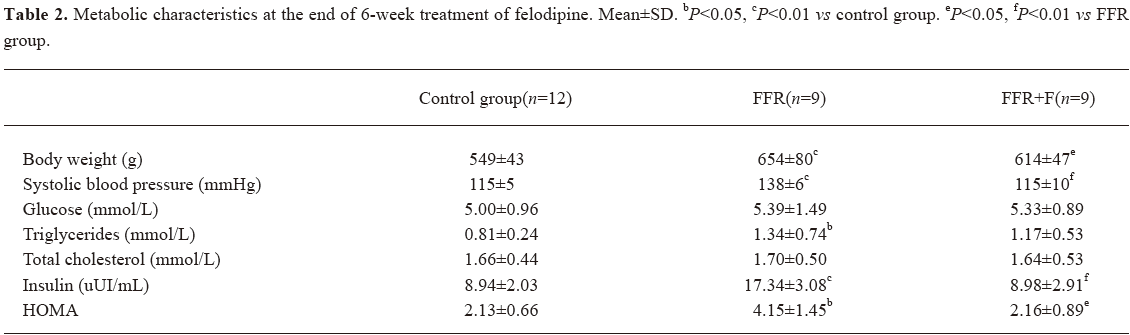
Full table
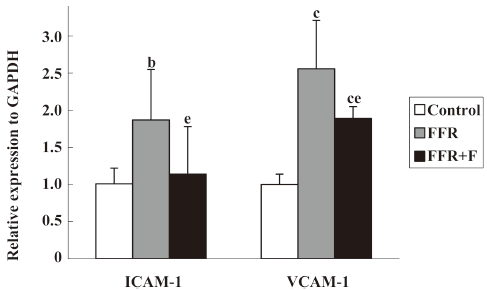
Effect of felodipine on adhesion molecules and macrophages in the aorta of FFR By real-time PCR, we found that the aortic expressions of ICAM-1 mRNA and VCAM-1 mRNA in FFR were 187%±68% (P<0.05) and 256%±56% (P<0.01) of that of the control group, respectively, after 38 weeks. After treatment with felodipine for 6 weeks, real-time PCR revealed that ICAM-1 mRNA was reduced by 39% (P=0.037) and VCAM-1 mRNA was reduced by 26% (P=0.020) when compared with FFR (Figure 1).
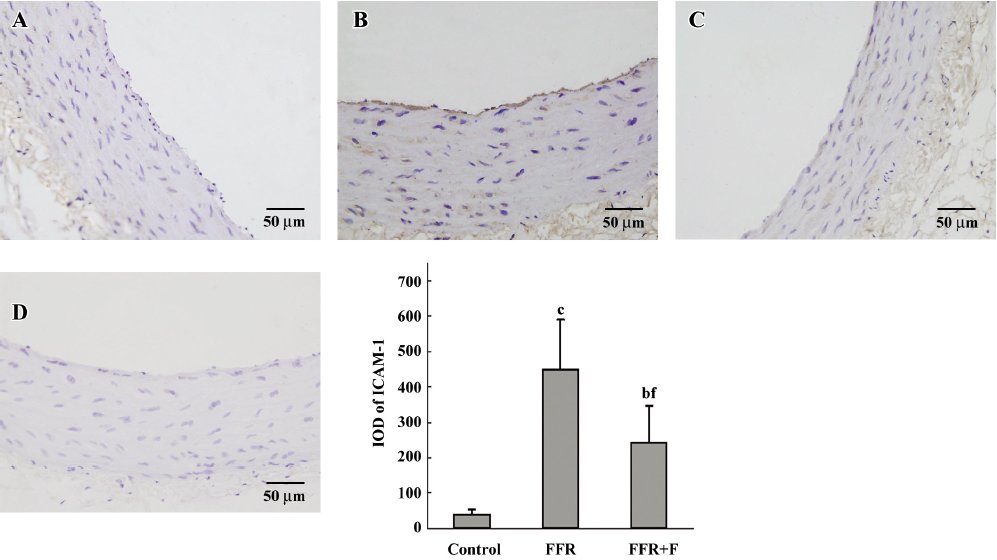
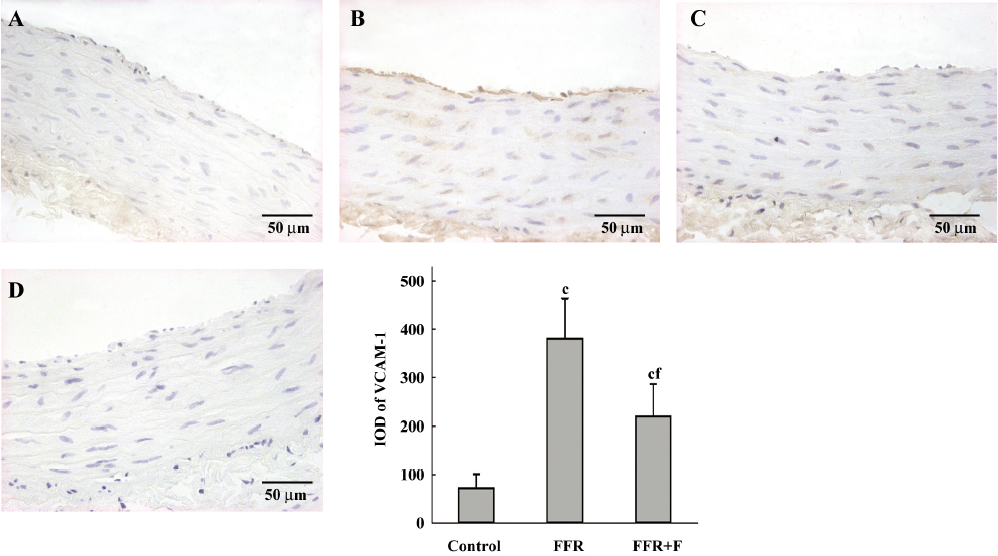
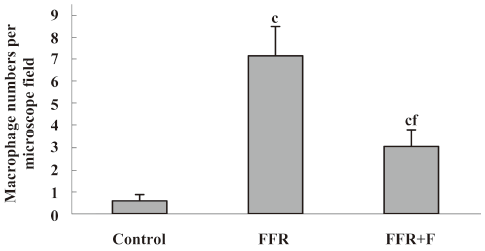
Immunohistochemistry was used to examine the localization of ICAM-1 and VCAM-1 in the aortic wall of FFR. ICAM-1 and VCAM-1 were most abundant in the intima of the aorta of FFR, and were weakly expressed in media. Felodipine reduced ICAM-1 and VCAM-1 expressions (Figures 2,3). Immunostaining with anti-CD68, a marker of monocytes/macrophages was enhanced in the aorta from FFR as compared with control rats (7.15±1.34 vs 0.59±0.27 cells/microscope field, P<0.01). Felodipine reduced the infiltration of macrophages significantly after the treatment (3.07±0.70 vs 7.15±1.34 cells/microscope field, P<0.01; Figure 4).
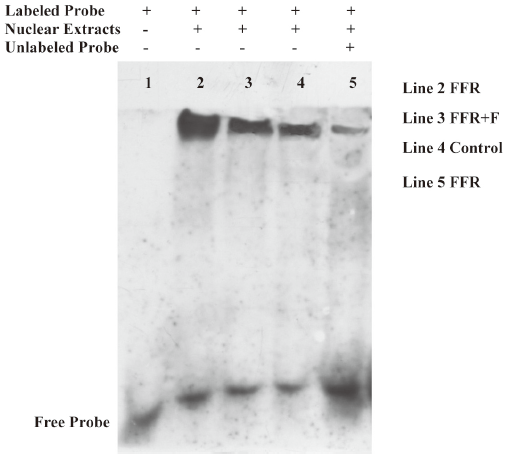
Effect of felodipine on NF-κB activity Because ICAM-1 and VCAM-1 gene expressions are regulated by NF-κB, we further examined the levels of NF-κB. Of importance, the binding activity of NF-κB, as assessed by the gel mobility shift assay, was markedly increased in the aorta of the FFR as compared with the normal rats, and the activation of NF-κB was markedly inhibited by treatment with felodipine for 6 weeks as compared with FFR (Figure 5).
Correlations between the levels of adhesion molecules and metabolic parameters As shown in Table 3, the expression of ICAM-1 mRNA correlated significantly with SBP and HOMA, and VCAM-1 mRNA correlated with SBP, triglycerides, insulin, and HOMA after 38 weeks.
Full table
In the subgroup analysis, we found that the expression of ICAM-1 mRNA correlated with levels of insulin (r=0.506, P<0.05), and VCAM-1 mRNA correlated with levels of insulin (r=0.495, P<0.05) and HOMA (r=0.509, P<0.05) in rats treated with felodipine. The levels of blood pressure, triglycerides, and cholesterol did not correlate with the adhesion molecules in the felodipine group.
Discussion
The present study demonstrates that ICAM-1 and VCAM-1 expressions and macrophage infiltration were enhanced in the aorta of high FFR, accompanied by the activation of NF-κB. This indicated that vascular inflammation existed in the aorta of rats with metabolic syndrome. Another striking finding was that felodipine, a CCB, reduced the expressions of ICAM-1 and VCAM-1 in this rat model of metabolic syndrome via the inhibition of NF-κB activity.
Evidence from both human[22] and animal[23] studies indicate that inflammatory mediators, such as interleukin-6 and hypersensitivity C-reaction protein (hsCRP), are involved in the pathogenesis of metabolic syndrome. Patients with metabolic syndrome are associated with increased atherosclerotic burden, as reflected by increased incidence of carotid[6] and coronary artery[5] plaques. It is well recognized that atherosclerosis is an inflammatory disease[8]. The expression of inducible adhesion molecules in the arterial wall represents a key event in the development of atherosclerosis. Adhesion molecules on the endothelial layer, such as ICAM-1 and VCAM-1, contribute to lesion initiation by mediating the recruitment of monocytes to the intima[24]. The recruited monocytes migrate into the subendothelial space and differentiate into macrophages[8]. Abundant macrophages containing plaques are vulnerable to fatal disruption in humans[25]. The present study shows that the ICAM-1 and VCAM-1 gene expressions were upregulated in the aorta in FFR, despite the absence of the aortic plaques. Immunohistochemistry showed the distinct expressions of ICAM-1 and VCAM-1 in the intima, and the infiltration of macrophages into the vascular wall was also enhanced. This expression pattern is similar to that in classical atherosclerosis[26]. Furthermore, we found that aortic adhesion molecular mRNA levels correlated with metabolic parameters, such as triglycerides, insulin, and HOMA. Thus increased adhesion molecular expression may participate in accelerating the growth of early lesions in patients with metabolic syndrome in a manner that is similar to the proposed role of adhesion molecules in the expansion of classical lesions. Given the special pathophysiological state[1,3], these data provide a partial explanation of the higher incidence of atherosclerosis in metabolic syndrome populations.
CCB are widely used to treat patients with hypertension and stable angina pectoris in clinical practice[12,13]. Clinical studies have shown that CCB retard the progression of atherosclerosis[12,13]. However, the underlying mechanisms remain unclear. Using ApoE-deficient mice, Yoshii et al[27] and Kataoka et al[28] found that amlodipine regresses atherosclerosis via anti-inflammatory action. Yao et al[29] demonstrated that felodipine suppresses atherosclerosis in high-cholesterol-fed ApoE-deficient mice by reducing oxidative stress and inflammatory cytokines. Because metabolic syndrome is characterized by the simultaneously occurring and clustering of several cardiovascular and metabolic risk factors[1,2], it is of great interest to investigate the effect of CCB on metabolic syndrome. To the best of our knowledge, this is the first study to demonstrate that felodipine decreases the expressions of ICAM-1 and VCAM-1 and reduces macrophage infiltration via the inhibition of NF-κB activity in the aorta of FFR. Our results suggest that felodipine may have the potential to inhibit the vascular inflammatory process in metabolic syndrome. In accordance with previous studies[27–29], our result expand our knowledge of retarding vascular damage in metabolic syndrome with the treatment of felodipine. Interestingly, we only found that the aortic expression of the adhesion molecules correlated with insulin and HOMA, but not with blood pressure and lipids after treatment with felodipine for 6 weeks. Clinical studies have demonstrated that CCB attenuate insulin resistance in hypertensive and diabetic patients[30,31]. In line with those studies, our results indicate that felodipine, a CCB, improves insulin resistance in FFR.
The expression of adhesion molecules is regulated transcriptionally by NF-κB[32]. NF-κB is a key transcription factor in vascular inflammation and is activated by pro-inflammatory cytokines, atherogenic lipoproteins, and reactive oxygen intermediates[33]. NF-κB is also involved in the development of insulin resistance[34]. Therefore, we suspect that NF-κB is involved in mediating the increased expression of adhesion molecules in the aorta of FFR because the above-mentioned stimuli are all related to metabolic syndrome[22,23]. Using EMSA, the most wildly applied method[35], we clearly demonstrated that the binding activity of NF-κB to its consensus oligonucleotide decreased after treatment with felodipine for 6 weeks in the aorta of FFR. Thus felodipine reduces the expression of adhesion molecules probably by inhibiting NF-κB activity in this rat model of metabolic syndrome.
As mentioned earlier, CCB have anti-inflammatory effects in atherosclerosis[27,28], and as inflammation is involved in metabolic syndrome[22,23], it is rational to consider that felodipine exerts its anti-inflammatory effects in this rat model of metabolic syndrome. However, the exact mechanism needs to be further elucidated.
Feeding fructose-enriched diets to normal rats has been shown to induce hypertension, insulin resistance and hyperinsulinemia, and provide a model of dietary-induced metabolic syndrome[14–17]. Dai et al[14] demonstrated that treatment with 10% fructose in drinking water (which is equivalent to a diet containing 48%–57% fructose) for at least 1 week is appropriate for the rapid induction of metabolic syndrome in Wistar rats. Takagawa et al[36] reported that the body weight of FFR was significantly increased after feeding a high fructose diet for 40 weeks, which suggests high fructose-induced metabolic syndrome is duration dependent. High fructose feeding failing to induce obesity may be one shortcoming of most studies using FFR, as obesity is now considered a major abnormality of metabolic syndrome, and this has been emphasized in the definition of metabolic syndrome by the International Diabetes Federation[37]. In our study, we used 10% fructose in drinking water to feed rats for 32 weeks. The FFR demonstrated increased body weight, hypertension, hyperinsulinemia, hyperglyceridemia, and insulin resistance, thus this model is similar to the characteristics of metabolic syndrome in humans.
Dietary and lifestyle changes reduce the cardiovascular risk in patients with metabolic syndrome. However, in those patients, the additional administration of antihypertensive drugs is required when there is hypertension. Because cardiovascular risk is high in hypertensive patients with metabolic syndrome, it would appear advisable to pursue a rigorous blood pressure control[38]. It is suggested that treating patients at high risk of cardiovascular disease with drugs that decrease inflammation improves outcome[39]. Our results may help us to well understand the beneficial effects of CCB in clinical practice.
The present study has some limitations. Other adhesion molecules, such as E-selectin and P-selectin, were not examined in our study. Our selection was based on the study which showed that only ICAM-1 and VCAM-1 were found at the sites predisposed to lesion formation in rabbit and mouse atherosclerosis[26]. The roles of other adhesion molecules in the progression of vascular damage in metabolic syndrome need to be further studied. Blood pressure was measured by tail-cuff methods in the present study. A recent publication[40] has shown that blood pressure does not increase in FFR when measured by radiotelemetry, and that the apparent increase of blood pressure observed using tail-cuff methods may be an artifact arising from the stress of using the animal restraint. In our study, efforts were made to minimize stress during blood pressure measurements, and our results are consistent with other studies that found blood pressure was elevated in FFR when measured by tail-cuff methods[14–17]. Considering that baseline blood pressure is not raised in all rats, and control rats and FFR experience the same level of stress in the tail-cuff procedure, it is reasonable to presume that fructose induces hypertension in rats.
In summary, the present study provides substantial evidence that vascular inflammatory responses are enhanced in FFR, a rat model of metabolic syndrome. The augmented inflammatory response in the arterial wall may be involved in the accelerated atherosclerosis in metabolic syndrome. Felodipine, a CCB, attenuates vascular inflammation through the inhibition of NF-κB activation, and may thus have the potential to inhibit vascular inflammation in metabolic syndrome.
References
- Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005;365:1415-28.
- Zhong M, Tan HW, Gong HP, Wang SF, Zhang Y, Zhang W. Increased serum visfatin in patients with metabolic syndrome and carotid atherosclerosis. Clin Endocrinol (Oxf) 2008. Epub ahead of print. [Crossref]
- van Zwieten PA, Mancia G. Background and treatment of metabolic syndrome: a therapeutic challenge. Semin Cardiothorac Vasc Anesth 2006;10:206-14.
- Gami AS, Witt BJ, Howard DE, Erwin PJ, Gami LA, Somers VK, et al. Metabolic syndrome and risk of incident cardiovascular events and death: a systematic review and meta-analysis of longitudinal studies. J Am Coll Cardiol 2007;49:403-14.
- Kullo IJ, Cassidy AE, Peyser PA, Turner ST, Sheedy PF II, Bielak LF. Association between metabolic syndrome and subclinical coronary atherosclerosis in asymptomatic adults. Am J Cardiol 2004;94:1554-8.
- Tzou WS, Douglas PS, Srinivasan SR, Bond MG, Tang R, Chen W, et al. Increased subclinical atherosclerosis in young adults with metabolic syndrome: the Bogalusa Heart Study. J Am Coll Cardiol 2005;46:457-63.
- Delbosc S, Paizanis E, Magous R, Araiz C, Dimo T, Cristol JP, et al. Involvement of oxidative stress and NADPH oxidase activation in the development of cardiovascular complications in a model of insulin resistance, the fructose-fed rat. Atherosclerosis 2005;179:43-9.
- Ross R. Atherosclerosis––an inflammatory disease. N Engl J Med 1999;340:115-26.
- Nageh MF, Sandberg ET, Marotti KR, Lin AH, Melchior EP, Bullard DC, et al. Deficiency of inflammatory cell adhesion molecules protects against atherosclerosis in mice. Arterioscler Thromb Vasc Biol 1997;17:1517-20.
- Benitez MB, Cuniberti L, Fornari MC, Rosso LG, Berardi V, Elikir G, et al. Endothelial and leukocyte adhesion molecules in primary hypertriglyceridemia. Atherosclerosis 2008;197:679-87.
- Haubner F, Lehle K, Munzel D, Schmid C, Birnbaum DE, Preuner JG. Hyperglycemia increases the levels of vascular cellular adhesion molecule-1 and monocyte-chemoattractant-protein-1 in the diabetic endothelial cell. Biochem Biophys Res Commun 2007;360:560-5.
- Nissen SE, Tuzcu EM, Libby P, Thompson PD, Ghali M, Garza D, et al. Effect of antihypertensive agents on cardiovascular events in patients with coronary disease and normal blood pressure: the CAMELOT study: a randomized controlled trial. JAMA 2004;292:2217-25.
- Pitt B, Byington RP, Furberg CD, Hunninghake DB, Mancini GB, Miller ME, et al. Effect of amlodipine on the progression of atherosclerosis and the occurrence of clinical events. PREVENT Investigators. Circulation 2000;102:1503-10.
- Dai S, McNeill JH. Fructose-induced hypertension in rats is concentration-and duration-dependent. J Pharmacol Toxicol Methods 1995;33:101-7.
- Zhao C, Wang P, Xiao X, Chao J, Chao L, Wang DW, et al. Gene therapy with human tissue kallikrein reduces hypertension and hyperinsulinemia in fructose-induced hypertensive rats. Hypertension 2003;42:1026-33.
- Tay A, Ozcelikay AT, Altan VM. Effects of L-arginine on blood pressure and metabolic changes in fructose-hypertensive rats. Am J Hypertens 2002;15:72-7.
- Miatello R, Vazquez M, Renna N, Cruzado M, Zumino AP, Risler N. Chronic administration of resveratrol prevents biochemical cardiovascular changes in fructose-fed rats. Am J Hypertens 2005;18:864-70.
- Barros CM, Lessa RQ, Grechi MP, Mouço TL, Souza MG, Wiernsperger N, et al. Substitution of drinking water by fructose solution induces hyperinsulinemia and hyperglycemia in hamsters. Clinics 2007;62:327-34.
- Vaziri ND, Wang XQ, Ni ZN, Kivlighn S, Shahinfar S. Effects of aging and AT-1 receptor blockade on NO synthase expression and renal function in SHR. Biochim Biophys Acta 2002;1592:153-61.
- Ortlepp JR, Breuer J, Eitner F, Kluge K, Kluge R, Floege J, et al. Inhibition of the renin-angiotensin system ameliorates genetically determined hyperinsulinemia. Eur J Pharmacol 2002;436:145-50.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods 2001;25:402-8.
- Brunner EJ, Hemingway H, Walker BR, Page M, Clarke P, Juneja M, et al. Adrenocortical, autonomic, and inflammatory causes of the metabolic syndrome: nested case-control study. Circulation 2002;106:2659-65.
- Xu ZG, Lanting L, Vaziri ND, Li Z, Sepassi L, Rodriguez-Iturbe B, et al. Upregulation of angiotensin II type 1 receptor, inflammatory mediators, and enzymes of arachidonate metabolism in obese Zucker rat kidney: reversal by angiotensin II type 1 receptor blockade. Circulation 2005;111:1962-9.
- Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis 2003;170:191-203.
- Moreno PR, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT. Macrophage infiltration in acute coronary syndromes. Implications for plaque rupture. Circulation 1994;90:775-8.
- Iiyama K, Hajra L, Iiyama M, Li H, DiChiara M, Medoff BD, et al. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res 1999;85:199-207.
- Yoshii T, Iwai M, Li Z, Chen R, Ide A, Fukunaga S, et al. Regression of atherosclerosis by amlodipine via anti-inflammatory and anti-oxidative stress actions. Hypertens Res 2006;29:457-66.
- Kataoka C, Egashira K, Ishibashi M, Inoue S, Ni W, Hiasa K, et al. Novel anti-inflammatory actions of amlodipine in a rat model of arteriosclerosis induced by long-term inhibition of nitric oxide synthesis. Am J Physiol Heart Circ Physiol 2004;286:H768-74.
- Yao R, Cheng X, Liao YH, Chen Y, Xie JJ, Yu X, et al. Molecular mechanisms of felodipine suppressing atherosclerosis in high-cholesterol-diet apolipoprotein E-knockout mice. J Cardiovasc Pharmacol 2008;51:188-95.
- Yagi S, Goto S, Yamamoto T, Kurihara S, Katayama S. Effect of cilnidipine on insulin sensitivity in patients with essential hypertension. Hypertens Res 2003;26:383-7.
- Ersoy C, Imamoðlu S, Budak F, Tuncel E, Ertürk E, Oral B. Effect of amlodipine on insulin resistance & tumor necrosis factor-alpha levels in hypertensive obese type 2 diabetic patients. Indian J Med Res 2004;120:481-8.
- Zhang WJ, Frei B. Alpha-lipoic acid inhibits TNF-alpha-induced NF-kappaB activation and adhesion molecule expression in human aortic endothelial cells. FASEB J 2001;15:2423-32.
- Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest 2001;107:255-64.
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 2005;11:191-8.
- van den Berg R, Haenen GR, van den Berg H, Bast A. Transcription factor NF-kappaB as a potential biomarker for oxidative stress. Br J Nutr 2001;86 Suppl 1:S121-7.
- Takagawa Y, Berger ME, Hori MT, Tuck ML, Golub MS. Long-term fructose feeding impairs vascular relaxation in rat mesenteric arteries. Am J Hypertens 2001;14:811-7.
- International Diabetes Federation. The IDF consensus world-wide definition of the metabolic syndrome. Available from URL: Metabolic syndrome definitionhttp://www.idf.org/webdata/docs/IDF
- Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, Germano G, et al. 2007 Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007;25:1105-87.
- Athyros VG, Kakafika AI, Karagiannis A, Mikhailidis DP. Do we need to consider inflammatory markers when we treat atherosclerotic disease? Atherosclerosis 2008. [Crossref]
- D’Angelo G, Elmarakby AA, Pollock DM, Stepp DW. Fructose feeding increases insulin resistance but not blood pressure in Sprague-Dawley rats. Hypertension 2005;46:806-11.