
Ginkgo biloba extract prevents against apoptosis induced by high glucose in human lens epithelial cells1
Introduction
In many countries cataracts are a major ocular complication of diabetes. At present, it affects approximately 60%–65% of type 2 diabetic patients, ultimately leading to blindness. Many studies suggest that people with diabetes are more predisposed to cortical and posterior subcapsular cataracts than those without diabetes, and those cataracts are characterized by low cell density as well as apoptosis[1–3]. This suggests that apoptosis in human lens epithelial cells (HLEC) is an initiating and key event in non-congenital cataracts, including diabetic cataract (DC) formation in humans and animals[4].
However, the exact mechanism underlying high glucose-induced HLEC apoptosis in DC remains unknown. There is now sufficient evidence implicating oxidative stress as a key factor in the etiology and progression of DC. High glucose is presumed to be an initiating agent, which induces oxidative stress and activates aldose reductase (AR)[5]. It is well known that oxidative stress plays an important role in the apoptosis of HLEC, and AR may be an essential mediator of cell death induced by high glucose. Furthermore, AR is also a significant regulator of the cellular redox state, which is sufficient to perturb the NADPH/NADP ratio and induce oxidative stress during hyperglycemia. Bcl-2 and Bax are Bcl-2 family members, which are considered to be the principal factors in determining whether the execution of apoptosis proceeds via the activation of caspases[6]. Caspases are key regulating enzymes in apoptosis in HLEC. Among them, caspase-3 has been identified as a key executor of apoptosis and is one of the most important caspases in the downstream of apoptotic pathways[7].
The tree Ginkgo biloba has long been believed to have medicinal properties. Its extracts are among the most widely sold herbal supplements in the world. Ginkgo biloba extract (GBE) used in our study is a standard extract of Ginkgo biloba, which is a defined complex mixture containing 24% Ginkgo flavone glycoside (quercetin, kaempferol, isorhamnetin) and 6% terpene lactones (ginkgolides, bilobalide). It has been used as a therapeutic agent in some cardiovascular and neurological disorders[8,9]. Although there are still few published reports that focus on the protection of GBE on DC, evidence accumulated in vitro and in vivo show that GBE can scavenge reactive oxygen species (ROS)[10,11] and suppress AR activity[12]. It also has pro-apoptotic actions in chemical stress[13]. All of these actions offer us a pharmacological foundation of GBE for DC therapy. Furthermore, our previous study demonstrated that GBE has a protective effect on cataracts in rats induced by streptozotocin (STZ). At the cellular level, however, the protective mechanisms of GBE on the apoptosis of DC have not been identified.
In our present work, using bendazac lysine (BDL), an original anticataract drug used as a control drug, we studied the possible influence of GBE on the expression of Bcl-2 and Bax, and the activities of caspase-3 in HLEC. These were all closely related to the mitochondrial signal pathway induced by oxidative stress and on HLEC apoptosis, which is the cellular basis for DC.
Materials and methods
Drugs HLEC line-B3 (Lot N
Lens epithelial cell culture and treatment The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; with 5.56 mmol/L glucose) including 20% fetal bovine serum (FBS), 100 IU/mL penicillin, 100 mg/mL streptomycin, and 2 mmol/L glutamine at 37 °C in a 5% CO2 humidified atmosphere. Cells at passages 20–27 were used for this study. Cells were grown in normal media until they were 60%–80% confluent. Cell growth was arrested for 12–24 h by replacing with fresh media containing 0.5% FBS and 50 µg/mL gentamicin. The low serum levels were maintained during growth arrest to prevent the slow apoptosis that accompanies complete serum deprivation. After 12–24 h, the cells were divided into 6 groups: normal glucose group (NS; 5.56 mmol/L glucose–DMEM), high glucose group (HG; 50 mmol/L glucose-DMEM), low-dose GBE group (GL; 10 µg/mL GBE–HG–DMEM), moderate-dose GBE group (GM; 20 µg/mL GBE–HG–DMEM), high-dose GBE group (GH; 40 µg/mL GBE–HG–DMEM), and the BDL group (0.5 µmol/mL BDL–HG–DMEM). Every group contained 6 bottles: After 24 h incubation, the cells were harvested and the mixture was centrifuged. The supernatants were then stored at –20 °C for later analysis.
Cell viability assay by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide colorimetry The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (the mononuclear cell direct cytotoxicity assay) was used as an additional index of cell viability. The growth-arrested cells were divided into 10 groups: the osmolarity control group (OG; 5.56 mmol/L glucose–DMEM with 45 mmol/L mannitol), NS group, HG group, different doses of the GBE groups (2, 5, 10, 20, 40, and 60 µg/mL GBE–HG–DMEM), and the BDL group. After the indicated treatments, 10 µL of 5 mg/mL MTT reagent (Sigma, St Louis, MO, USA) was added to each well of a 96-well microplate, and incubated in the dark at 37 °C for 4 h. Finally, 200 µL DMSO (Sigma, USA) was added as the MTT formazan-product solvent to each well with vigorous mixing after the supernatant was removed. Afterwards, the optical density (OD) at 490 nm was measured with a microplate reader (BMG, Germany). Cell viability was expressed as a percentage of the 5.56 mmol/L condition.
Apoptotic analysis by flow cytometry in HLEC The percentage of apoptotic cells was determined by cell flow cytometry, which was conducted according to the protocol outlined by the manufacturer of the Annexin V–FITC apoptosis detection kit. Briefly, after the corresponding treatment, the cells were harvested with 0.25% trypsin and washed with cold phosphate-buffered saline (PBS) at 5000×g at 4 °C for 5 min 3 times. The cell pellets were resuspended in binding buffer (1×) prior to the addition of FITC-labeled Annexin V for 15 min at room temperature in the dark. Propidium iodide (PI) was added at a final concentration of 2 µg/mL, and cell apoptosis was immediately analyzed on a flow cytometer (Becton Dickinson, USA). Apoptotic cells were defined as FITC+/PI- cells.
Analysis of nuclear morphology in HLEC After the indicated treatments, HLEC incubated on 6-well culture plates were washed with cold PBS for 3 min 3 times, fixed by 4% paraformaldehyde for 15 min, washed with cold PBS, and incubated with 10 µg/mL Hoechst 33258, a DNA-binding fluorescent dye, for 10 min. The cells were examined under a fluorescent microscope (Olympus, Tokyo, Japan) using an excitation wavelength of 350 nm and an emission wavelength of 460 nm. Apoptotic cells were defined on the basis of nuclear morphological changes, such as chromatin condensation, and fragmentation.
Measurement of oxidative stress in HLEC Total antioxidative capability (T-AOC), the activities of catalase (CAT), total superoxidase dismutase (T-SOD), and glutathione–peroxidase (GSH-Px) in HLEC were measured by spectrophotometry using kits from Jiancheng Bioengineering Institute (Lot N
Measurement of AR activity in HLEC[14–16] The activities of AR in the cytosolic fraction were estimated spectrophotometrically. Harvested HLEC were ultrasonically disrupted in 100 mmol/L PBS (pH 6.8), followed by centrifugation at 25 000×g for 5 min. The supernatants were assumed to be the crude AR enzyme fraction. The reaction was carried out in 1.5 mL incubation medium (100 mmol/L PBS, pH 6.8, 0.1 mmol/L DL-glyceraldehyde, 0.15 mmol/L NADPH, and 100 µL crude AR enzyme fraction); DL-glyceraldehyde was added last. Enzyme activity was measured spectrophotometrically by estimating NADPH oxidation from a decrease in the OD at 340 nm. Assays were carried out at room temperature with an appropriate blank subtracted from each reaction to correct for non-specific oxidation of NADPH during the measurement. Its activity was defined as the amount of enzymes catalyzing the oxidation of 1 µmol NADPH per minute under the assay conditions. The protein concentration was determined by the Bradford method at the Beyotime Institute of Biotechnology (China).
Determination of Bcl-2 and Bax by Western blot analysis in HLEC The HLEC were seeded onto 6-well plates with 2×105 cells/mL and given the indicated treatment. After removal of the media, cells were washed twice with ice-cold PBS, then lysed using cell lysis buffer (20 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L EDTA, 1% Na3VO4, 0.5 µg/mL leupeptin, and 1 mmol/L phenylmethanesulfonyl fluoride) for 20 min on ice. The lysates were collected by scraping from the plates, and then centrifuged at 12 000×g at 4 °C for 5 min. Briefly, the proteins (50 µg) were loaded on a 12.5% of SDS–polyacrylamide gel for electrophoresis, then transferred onto nitrocellulose transfer membranes (Amersham Biosciences, Buckinghamshire, UK) at 0.8 mA/cm2 for 30 min. The membranes were blocked at room temperature for 1 h with blocking solution [5% skimmed milk in Tris-buffered solution plus Tween-20 (TBST): 50 mmol/L Tris-HCl, 150 mmol/L NaCl, pH7.5, and 0.1% v/v Tween-20]. The membranes were then incubated overnight at 4 °C with an anti-Bcl-2 or anti-Bax rabbit polyclonal antibody at 1:200 in blocking solution. After washing with TBST, the membranes were incubated for 2 h at room temperature with a horseradish peroxidase-conjugated antirabbit secondary antibody at 1:5000 in blocking solution. NBT/BCIP (Promega, Madison, WI, USA) was used as a color substrate to develop the membranes. The densities of the bands on the membranes were scanned and analyzed with an image analyzer (Quantity One; Bio-Rad, USA). β-actin of HLEC was used as a housekeeping protein and was determined following the same procedure as mentioned earlier using a specific anti-actin rabbit polyclonal antibody (Sigma–Aldrich, Madrid, Spain) at a dilution of 1:1000 in blocking solution and a horseradish peroxidase-conjugated antimouse secondary antibody at a dilution of 1:500 in blocking solution.
Determination of caspase-3 activities Caspase-3 activities were determined by a colorimetric assay based on the ability of caspase-3 to change acetyl-Asp-Glu-Val-Asp p-nitroanalide into a yellow formazan product (p-nitroanalide). An increase in absorbance at 405 nm was used to quantify caspase-3 activities. After the corresponding treatment, HLEC were collected and rinsed with cold PBS, and then lysed by lysis buffer for 15 min on ice. Cell lysates were centrifuged at 18 000×g for 10 min at 4 °C. The enzymatic reaction for caspase-3 activities was carried out on a 96-well microplate with 35 µL cell lysate, 50 µL reaction buffer containing 5 µmol/L dithiothreitol, and 5 µL of 1 mmol/L caspase-3 colorimetric substrate at 37 °C for 4 h. The OD was determined at 405 nm using the 3550-UV microplate reader. The OD was obtained from a reaction in which no substrate was added, and was subtracted from each value, that is OD405. Exact proteins of every sample were assayed by Coomassie brilliant blue colorimetry at 595 nm. The OD405/OD595 was expressed as the relative activity of caspase-3. All the experiments were carried out in triplicate.
Statistical analysis Statistical analysis was performed to compare the effects of GBE on HLEC by one-way ANOVA followed by the least significant difference test or Dunnett’s t-test (2-sided) for different groups using SPSS 13.0 software (SPSS, Chicago, IL, USA). The data were presented as mean values and standard deviations. The number of samples in each group was 6. P<0.05 was considered statistically significant.
Results
Effect of GBE on the cell viability of HLEC The cell viability in the HG group was greatly lower than that of the NS group (P<0.01), but compared with that of the NS group, the cell viability in the OG group had no significant change (P>0.05). It suggested that osmolarity had no evident effect on HLEC. These results supported the findings of a similar study[17,18]. Compared with the HG group, the 10–40 µg/mL GBE groups increased HLEC viability in a significant, dose-dependent manner. The protection of HLEC cultured in high glucose was not significant in the 2 and 5 µg/mL of GBE group (P>0.05). Compared with the 20 µg/mL GBE group, the action of protecting HLEC was not obvious in the 60 µg/mL GBE group (P>0.05). The 40–60 µg/mL GBE groups had protective effects on HLEC, but had no significant dose dependence. Therefore, we used 10, 20, and 40 µg/mL GBE as our investigative dosage in our later study (Table 1).
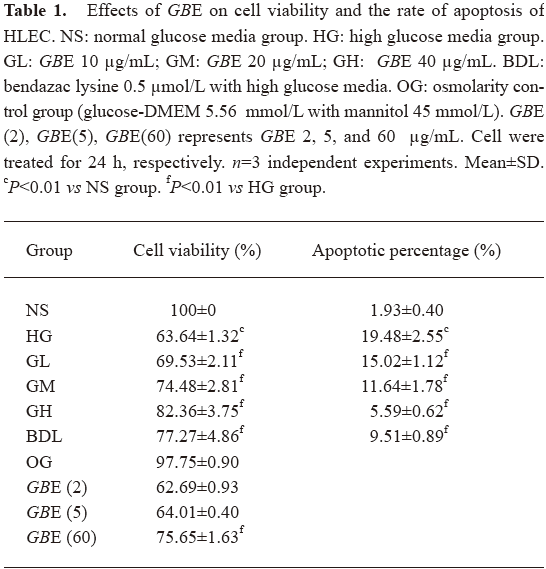
Full table
Effect of GBE on the relative rate of HLEC apoptosis Annexin V+ and PI- cells were designated as apoptotic cells. Table 1 shows that the number of apoptotic cells in the HG group was higher than that of the NS group, indicating that high glucose led to the translocation of phosphatidylserine, an early event of the apoptotic process. Compared to the HG group, a significant decrease in apoptotic cells was observed in the GL, GM, GH, and BDL groups (P<0.01; Table 1).
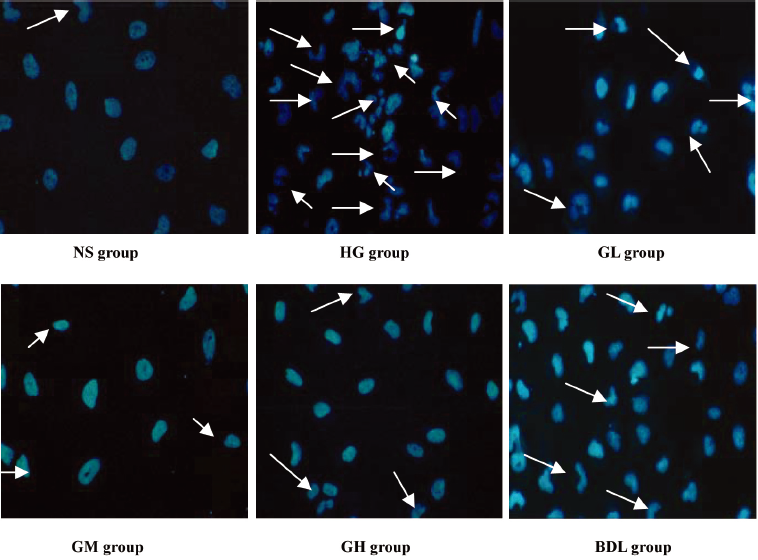
Effect of GBE on morphological characterization of HLEC apoptosis Apoptosis was further confirmed by analyzing the nuclear morphology of HLEC. Nuclear morphology was evaluated with membrane-permeable blue Hoechst33258. Figure 1 shows Hoechst 33258 fluorescence photomicrographs of cultured HLEC. In the NS group, the nuclei of HLEC had regular contours and were round and large in size. HLEC with smaller nuclei and condensed chromatin were rarely seen. In contrast, most nuclei of high glucose-treated HLEC appeared to be hypercondensed. The number of apoptotic nuclei containing condensed chromatin decreased gradually in the GL, GM, GH, and BDL groups compared to the HG group (Figure 1).
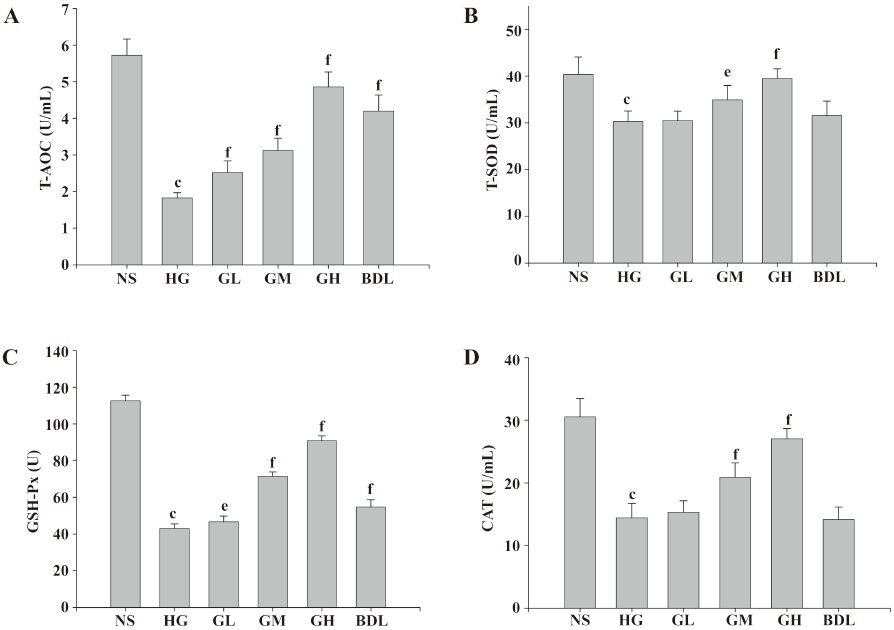
Effects of GBE on oxidative stress in HLEC The activities of the CAT, GSH-Px, and T-SOD, and the level of T-AOC in the HG group were all significantly lower than those of the NS group (P<0.05 or P<0.01), suggesting that HLEC in high glucose exhibited distinct oxidative stress. It was also found that the 3 antioxidase activities and the level of T-AOC were dramatically raised in the GM and GH groups (P<0.05 or P<0.01), but BDL had no evident effect on T-AOC and T-SOD, and low-dose GBE had also no evident effects on T-SOD and CAT (P>0.05; Figure 2).
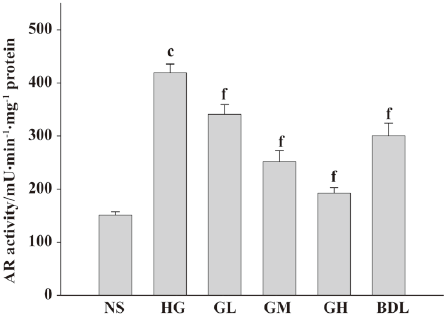
Effect of GBE on AR activity in cultured HLEC The AR activity of the HG group was dramatically increased (P<0.01), suggesting that high glucose can activate AR greatly. When compared with the HG group, AR relative activity in the GL, GM, and GH groups decreased significantly, respectively (P<0.01), suggesting that GBE inhibits AR activation induced by high glucose in a dose-dependent manner (Figure 3).
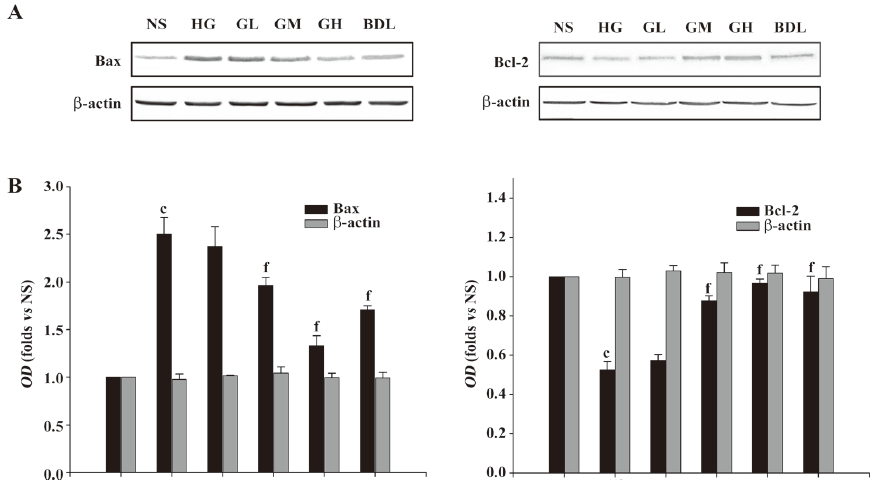
Effect of GBE on the expressions of Bcl-2 and Bax proteins As shown in Figure 4, the Bax proteins in the HG group greatly increased, while the Bcl-2 proteins markedly decreased compared to the NS group. The ratio of Bax to Bcl-2 in the HG group was significantly higher than that in the NS group (P<0.01). When the concentration of GBE increased, the ratio of Bax to Bcl-2 in the GL, GM, and GH groups significantly decreased (P<0.05 or P<0.01). The expressions of Bax and Bcl-2 in the BDL group had similar changes (P<0.01; Figure 4).
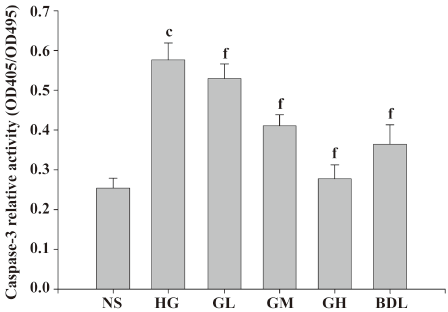
Effect of GBE on caspase-3 activity in HLEC Under high glucose (50 mmol/L) exposure, caspase-3 activities significantly increased compared to the NS group (P<0.01), suggesting that high glucose activated caspase-3 of HLEC dramatically. The caspase-3 activities of the GL, GM and GH groups were higher than those of the NS group in a dose-dependent manner (Figure 5).
Discussion
HLEC are the most metabolically-active part of the lens and are responsible for maintaining its homeostasis and transparency. Being the most anterior part of the lens, it is the primary site of external insult that ultimately leads to cataracts[19]. Cataracts are a multifactorial disease associated with several risk factors, of which diabetes is one of the most significant[20]. It is well known that the incidence of cataracts in human diabetic mellitus (DM) is significantly higher than that in non-diabetics[21]. In addition to the higher incidence, the pace of opacity development as a function of time is also faster in DM[22]. It is generally accepted that the apoptosis of HLEC is associated with the development of cataracts. High glucose is presumed to be an initiating agent and is regarded as a major cause of lens opacity. Our study was undertaken to examine the effects of GBE on high glucose-induced apoptosis of HLEC and to determine the possible molecular mechanisms involved.
In our present study, we found that in high glucose, the rate of apoptosis was increased and HLEC appeared to have typical apoptotic nuclei, such as condensed chromatin and nuclear fragmentation, but cell viability was decreased. All of these results suggest that high glucose as a stress factor induced HLEC apoptosis, which is consistent with previous reports[18]. When the cells were cocultured with GBE, all of the earlier indexes were significantly ameliorated correspondingly in a dose-dependent manner. These results demonstrate that GBE can strikingly inhibit cell apoptosis induced by high glucose and has potent cytoprotective action. However, little is known about the exact mechanism of the anti-apoptosis of GBE, so it would be worthwhile for us to further investigate.
It is clear that high glucose affects several stages in apoptotic signaling, and in most cell types studied, results in cell death by increasing oxidative and nitrosative stresses, activating pro-apoptotic Bcl-2 family proteins, and initiating a caspase cascade[23]. Oxidative stress has been known to play a critical role in the pathogenesis of cataracts with DM and is an important apoptotic signal. Considerable evidence shows that hyperglycemia would not only generate more ROS, but also attenuate endogenous antioxidative mechanisms through the glycation of scavenging enzymes and depletion of low molecular antioxidants. Shifts in redox balances contribute to the overt oxidative stress in diabetic individuals[24]. It activates protein kinase C (PKC), c-Jun-NH2-terminal kinase (JNK), and p38 mitogen-activated protein kinase (p38 MAPK), which lead to the activation of redox-sensitive transcription factors like NF-κB) and activated protein-1 (AP-1)[18]. Various antioxidants inhibit HLEC injury from high glucose and ameliorate the features of DC. It has been reported that the treatment of α-lipoic acid, vitamin C and vitamin E delay the development and progression of cataracts in rats with STZ-induced diabetes[25,26]. Therefore, antioxidant treatment is a potential therapy for DC. In our study, the activities of T-SOD, CAT, and GSH-Px, and the level of T-AOC (common indicators of changes in the anti-oxidation system) in high glucose were significantly decreased, suggesting that HLEC in high glucose exhibited oxidative stress. After the cells were incubated with GBE, the activities of T-SOD, CAT, and GSH-Px, and the level of T-AOC, all increased significantly, strongly suggesting that GBE has a potent antioxidative capability in vitro. Similar result for high glucose mesangial cells[11] and DN rats induced by STZ[10,27] were also found. The control drug BDL also had significant effects on enhancing GSH-Px activities and the T-AOC level, but there were no significant effects on CAT and T-SOD. These data suggest that GBE may have a more profound effect than BDL on ameliorating the oxidative stress state of HLEC.
AR is a monomeric-reduced, NADPH-dependent enzyme and a member of aldo-keto reductase superfamily. It is described as having glucose-reducing activity and is the key enzyme in the polyol pathway. The activation of AR in the polyol pathway is the early and critical event in high glucose-induced cell death[23]. The activation of this pathway could lead to the depletion of NADPH, thereby perturbing the cellular redox poise, leading to the formation of oxidative stress. It was found that AR was the major key enzyme for diabetes-induced oxidative stress in the lens[28] and its activity was essential for the apoptotic signaling events associated with high glucose. It was reported that the inhibition of AR prevented the activation of cellular kinases JNK, p38 MAPK, PKC, and redox-sensitive transcription factors like NF-κB and AP-1 in lens epithelial cells with high glucose culture[18]. Therefore, the inhibition of AR can ameliorate oxidative stress, and apoptosis in high glucose and delay the development of cataracts with DM. In our study, AR activities of the cells incubated in high glucose increased significantly, and decreased dramatically when incubated with GBE. This result strongly suggests that GBE has a potent inhibitory effect on the activation of AR. A similar effect of GBE was found on bovine lens[26]. Oxidative stress has been suggested to be a causative factor in the development of diabetic and hyperglycemic injury, and AR is the last and most common point in the oxidation–reduction pathway. The activation of AR can generate oxidative stress by perturbing redox poise, thus, our results may be important for understanding diabetic complications and beneficial for future treatments.
It has been found that the ratio of pro-apoptotic to anti-apoptotic Bcl-2-like proteins, which can regulate the mitochondrial cell death pathway, is altered in high glucose in a way that favors apoptosis[23]. Among them, Bcl-2 is an important anti-apoptotic proto-oncogene and Bax is a pro-apoptotic one. The ratio of increased Bax/Bcl-2 damages the integrity of mitochondria, causing the release of certain apoptosis-inducing proteins (eg cytochrome c and apoptosis-inducing factor) leading to the activation of caspase-3 via caspase-9[29]. Some or all of these mechanisms are involved in high glucose-induced apoptosis depending on the cell type or tissue studied[23]. It was reported that in renal tubular epithelial cells, high glucose caused a 2-fold increase in the Bax expression and a decrease in Bcl-2 expression[30]. Is there a similar effect on HLEC in high glucose? In our study, we found similar results. While the expression of Bcl-2 increased and that of Bax decreased, the ratio of Bax to Bcl-2 decreased significantly in HLEC incubated with GBE. These changes can inhibit apoptosis induced by high glucose. Therefore, in this aspect, GBE can be a prime candidate for the prevention and treatment of DC.
Cysteine aspartases (caspases), a protease family, are required for apoptosis induced by various stimuli[21]. Previous studies have demonstrated that caspase-3 plays a key role in the HLEC apoptosis induced by oxidative stress[18,23,31], suggesting that a mitochondria-mediated signaling pathway is involved. In mammals, caspase-3 is thought to be the main effector of caspases and has been identified as being activated in response to oxidative stress. The activation of caspase-3 is an important step in the execution phase of apoptosis and its inhibition blocks cell apoptosis[32]. In our study, we found that caspase-3 activity increased in high glucose, while in cells with GBE treatment, it decreased significantly, suggesting that high glucose-induced apoptosis was required for the caspase-dependent pathway in HLEC. The inhibitory effects of GBE on cell apoptosis involved inhibition of caspase-3 activation.
Mitochondria play an important role in apoptosis under a variety of pro-apoptotic conditions, such as oxidative stress[33]. The initiation of stress-induced apoptosis is much less defined, but seems to involve mitochondria at a very early stage of the intracellular signalling cascade[34]. High glucose makes HLEC exhibit oxidative stress, which may activate p53[35], JNK, p38 MAPK, etc[18], and then change the ratio of pro-apoptotic (Bax) to anti-apoptotic (Bcl-2) members, which is a major checkpoint in the common portion of this pathway[36], or change the mitochondrial membrane potential. All of these changes lead to the opening of the permeability transition pore, and the release of the intermembrane space protein, cytochrome c. Released cytochrome c activates Apaf-1, which in turn activates caspase-3 via caspase-9, ultimately leading to HLEC apoptosis. GBE can ameliorate the oxidative stress state, suppress AR and caspase-3 activation, and lower the ratio of Bax to Bcl-2 in cultured HLEC. Therefore, GBE can inhibit the HLEC intrinsic mitochondrial cell death pathway mediated by oxidative stress induced by high glucose on several pharmacological targets. Accordingly, this means that GBE may vitally postpone DC.
References
- Leske MC, Wu SY, Hennis A, Connell AM, Hyman L, Schachat A. Diabetes, hypertension, and central obesity as cataract risk factors in a black population. The Barbados Eye Study. Ophthalmology 1999;106:35-41.
- Klein BE, Klein R, Lee KE. Diabetes, cardiovascular disease, selected cardiovascular disease risk factors, and the 5-year incidence of age-related cataract and progression of lens opacities: the Beaver Dam Eye Study. Am J Ophthalmol 1998;126:782-90.
- Oishi N, Morikubo S, Takamura Y, Kubo E, Tsuzuki S, Tanimoto T, et al. Correlation between adult diabetic cataracts and red blood cell aldose reductase levels. Invest Ophthalmol Vis Sci 2006;47:2061-4.
- Li WC, Kuszak JR, Dunn K, Wang RR, Ma W, Wang GM, et al. Lens epithelial cell apoptosis appears to be a common cellular basis for non-congenital cataract development in humans and animals. J Cell Biol 1995;130:169-81.
- Pladzyk A, Ramana KV, Ansari NH, Srivastava SK. Aldose reductase prevents aldehyde toxicity in cultured human lens epithelial cells. Exp Eye Res 2006;83:408-16.
- Carambula SF, Matikainen T, Lynch MP, Flavell RA, Goncalves PB, Tilly JL, et al. Caspase-3 is a pivotal mediator of apoptosis during regression of the ovarian corpus luteum. Endocrinology 2002;143:1495-501.
- Qi JP, Wu AP, Wang DS, Wang LF, Li SX, Xu FL. Correlation between neuronal injury and caspase-3 after focal ischemia in human hippocampus. Chin Med J (Engl) 2004;117:1507-12.
- Siegel G, Schafer P, Winkler K, Malmsten M. Ginkgo biloba (EGb 761) in arteriosclerosis prophylaxis. Wien Med Wochenschr 2007;157:288-94.
- Zou W, Kim BO, Zhou BY, Liu Y, Messing A, He JJ. Protection against human immunodeficiency virus type 1 Tat neurotoxicity by Ginkgo biloba extract EGb 761 involving glial fibrillary acidic protein. Am J Pathol 2007;171:1923-35.
- Lu Q, Yin XX, Wang JY, Gao YY, Pan YM. Effects of Ginkgo biloba on prevention of development of experimental diabetic nephropathy in rats. Acta Pharmacol Sin 2007;28:818-28.
- Wang JY, Yin XX, Wu YM, Tang DQ, Gao YY, Wan MR, et al. Ginkgo biloba extract suppresses hypertrophy and extracellular matrix accumulation in rat mesangial cells. Acta Pharmacol Sin 2006;27:1222-30.
- Head KA. Natural therapies for ocular disorders, part two: cataracts and glaucoma. Altern Med Rev 2001;6:141-66.
- Thiagarajan G, Chandani S, Harinarayana Rao S, Samuni AM, Chandrasekaran K, Balasubramanian D. Molecular and cellular assessment of Ginkgo biloba extract as a possible ophthalmic drug. Exp Eye Res 2002;75:421-30.
- Srivastava S, Tammali R, Chandra D, Greer DA, Ramana KV, Bhatnagar A, et al. Regulation of lens aldose reductase activity by nitric oxide. Exp Eye Res 2005;81:664-72.
- Preet A, Siddiqui MR, Taha A, Badhai J, Hussain ME, Yadava PK, et al. Long-term effect of Trigonella foenum graecum and its combination with sodium orthovanadate in preventing histopathological and biochemical abnormalities in diabetic rat ocular tissues. Mol Cell Biochem 2006;289:137-47.
- Seo HG, Nishinaka T, Yabe-Nishimura C. Nitric oxide up-regulates aldose reductase expression in rat vascular smooth muscle cells: a potential role for aldose reductase in vascular remodeling. Mol Pharmacol 2000;57:709-17.
- Srivastava SK, Bhatnagar A, Friedrich B, Ramana KV. NADPH oxidase and aldose reductase inhibition attenuates high glucose-induced NF–kappaB activation and apoptosis of human lens epithelial cells. Invest Ophthalmol Vis Sci 2004;45:1698.
- Ramana KV, Friedrich B, Bhatnagar A, Srivastava SK. Aldose reductase mediates cytotoxic signals of hyperglycemia and TNF-alpha in human lens epithelial cells. FASEB J 2003;17:315-7.
- Mohanty I, Joshi S, Trivedi D, Srivastava S, Gupta SK. Lycopene prevents sugar-induced morphological changes and modulates antioxidant status of human lens epithelial cells. Br J Nutr 2002;88:347-54.
- Mulhern ML, Madson CJ, Danford A, Ikesugi K, Kador PF, Shinohara T. The unfolded protein response in lens epithelial cells from galactosemic rat lenses. Invest Ophthalmol Vis Sci 2006;47:3951-9.
- Biswas S, Harris F, Singh J, Phoenix D. Role of calpains in diabetes mellitus-induced cataractogenesis: a mini review. Mol Cell Biochem 2004;261:151-9.
- Hegde KR, Varma SD. Cataracts in experimentally diabetic mouse: morphological and apoptotic changes. Diabetes Obes Metab 2005;7:200-4.
- Allen DA, Yaqoob MM, Harwood SM. Mechanisms of high glucose-induced apoptosis and its relationship to diabetic complications. J Nutr Biochem 2005;16:705-13.
- Kyselova Z, Stefek M, Bauer V. Pharmacological prevention of diabetic cataract. J Diabetes Complications 2004;18:129-40.
- Kojima M, Sun L, Hata I, Sakamoto Y, Sasaki H, Sasaki K. Efficacy of alpha-lipoic acid against diabetic cataract in rat. Jpn J Ophthalmol 2007;51:10-3.
- Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J 1999;13:23-30.
- Yin XX, Zhang YD, Liu X. The inhibitory effects of Ginkgo biloba extract and Astragalus saponin I on aldose reductase extracted from bovine lens. Acta Acad Med Xuzhou 2006;26:478-81.
- Chung SS, Ho EC, Lam KS, Chung SK. Contribution of polyol pathway to diabetes-induced oxidative stress. J Am Soc Nephrol 2003;14:S233-6.
- Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol 2000;10:369-77.
- Verzola D, Bertolotto MB, Villaggio B, Ottonello L, Dallegri F, Frumento G, et al. Taurine prevents apoptosis induced by high ambient glucose in human tubule renal cells. J Investig Med 2002;50:443-51.
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 2002;23:599-622.
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 1999;15:269-90.
- Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309-12.
- Gulbins E, Dreschers S, Bock J. Role of mitochondria in apoptosis. Exp Physiol 2003;88:85-90.
- Vousden KH. Outcomes of p53 activation––spoilt for choice. J Cell Sci 2006;119:5015-20.
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev 1999;13:1899-911.