
All-trans retinoic acid inhibits the increases in fibronectin and PAI-1 induced by TGF-β1 and Ang II in rat mesangial cells1
Introduction
Glomerulosclerosis is characterized by excessive deposition of extracellular matrix (ECM) in glomeruli. It represents a final common pathway leading to renal dysfunction in a variety of primary and secondary glomerular diseases such as diabetic nephropathy, lupus nephritis and chronic glomerulonephritis[1]. Glomerular mesangial cells are the major cells to produce mesangial ECM. Apart from increased synthesis of matrix protein, decreased degradation contributes to ECM buildup. ECM is mainly degraded via two distinct pathways: the matrix metalloproteinases (MMPs) degrading pathway and the plasminogen activators (PA)/plasmin proteolytic axis[2]. Plasmin is involved in ECM turnover by a direct promotion on the degradation of matrix proteins and by an indirect action on the activation of latent MMP. Inactive plasminogen is converted to active plasmin by tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA). Therefore, PAI-1, as a major inhibitor of t-PA and u-PA, is thought to account for ECM accumulation by inhibiting plasmin- and MMP-mediated ECM degradation. ECM degradation has been shown to be inhibited by plasmin inhibitors, and increased by PAI-1 monoclonal antibody in cultured mesangial cells[2]. PAI-1 level is very low and even undetectable in normal kidneys, but is dramatically elevated in various forms of kidney diseases[3]. A significant increase in renal fibrosis is found in transgenic mice overexpressing PAI-1 that are subjected to unilateral ureteral obstruction (UUO)[4]. In contrast, PAI-1-deficient mice have substantially less fibrosis in the kidney compared with wild-type mice in response to renal injury[5–7]. Furthermore, PAI-1 blockage using a nonfunctional PAI-1 competitor reduces pathological ECM accumulation by restoring plasmin generation and increasing plasmin-dependent degradation of matrix components in experimental glomerulonephritis and in cultured mesangial cells[8,9]. These studies strongly suggest an important role of PAI-1 in the pathogenesis of renal fibrosis. Transforming growth factor-β1 (TGF-β1) and angiotensin II (Ang II), identified as the key fibrogenic cytokines, have been shown to be stimulators of ECM protein synthesis and potent inducers of PAI-1[10–15].
Retinoic acid (RA) is a group of derivatives of vitamin A (retinol), including all-trans RA (atRA), 9-cis RA and 13-cis RA. The action of RA is mediated by its receptors, which are ligand-dependent transcription factors that belong to the steroid/thyroid/vitamin D nuclear receptor superfamily. Two subfamilies of RA receptors with different ligand specificities are found, namely retinoic acid receptor (RAR) and retinoid X receptor (RXR), each of them has three isotypes (-α, -β, and -γ). RARs can be stimulated by atRA and 9-cis RA, RXRs are exclusively activated by 9-cis RA. RA and its receptor agonists have been demonstrated to protect the kidney in terms of renal structure and function in numerous animal models of renal diseases[16–20]. This beneficial effect of RA is ascribed to its anti-inflammatory and anti-proliferative properties. Moreover, previous study from our laboratory has shown that atRA effectively decreases cardiac fibrosis in spontaneously hypertensive rats[21]. However, the effect of atRA on the expression of fibronectin and PAI-1 in mesangial cells remains unknown. In the present study, we investigated the effect of atRA on the increases in PAI-1 and fibronectin induced by TGF-β1 and Ang II in cultured rat glomerular mesangial cells.
Materials and methods
Cell culture and treatment protocols Rat mesangial cells were kindly provided by Professor Guo MY (Shanghai Medical Center, Fudan University, Shanghai). Experiments were conducted with cells within passages 10. The cells were grown to 50%-60% confluence in Dulbecco’s modified Eagle’s medium (DMEM) (D-glucose concentration 5.6 mmol/L) (Sigma) supplemented with 10% fetal bovine serum (FBS) (Invitrogen) in a humidified atmosphere (5% CO2 and 95% air) at 37 °C. Before each experiment, cells were rendered quiescent by culturing in medium containing 1% FBS. 48 h later, cells were pretreated with atRA (Sigma, Lot No.043k1341 with purity of 98% by HPLC) at the indicated concentrations, and then stimulated with 2 μg/L TGF-β1 (R&D Systems Inc) or 0.1 μmol/L Ang II (Sigma) for the indicated periods of time. atRA was prepared and added to cells under reduced lighting conditions to limit spontaneous isomerization to 9-cis RA and 13-cis RA.
RT-PCR Total cellular RNA was extracted from mesangial cells with TRIzol according to the procedure recommended by the manufacturer (Shenergy Biocolor Co, Shanghai, China). 2 μg of total RNA was reversely transcribed to cDNA using oligodT (Shenergy Biocolor Co) and MMLV reverse transcriptase (Takara, Tokyo, Japan). cDNA was subjected to PCR amplification using specific primers and Taq DNA polymerase (Takara). PCR products were run on agarose gels and visualized by ethidium bromide staining. The DNA ladder was used as a standard. The primers, experimentally determined optimal annealing temperature and expected size of PCR products are presented in Table 1.
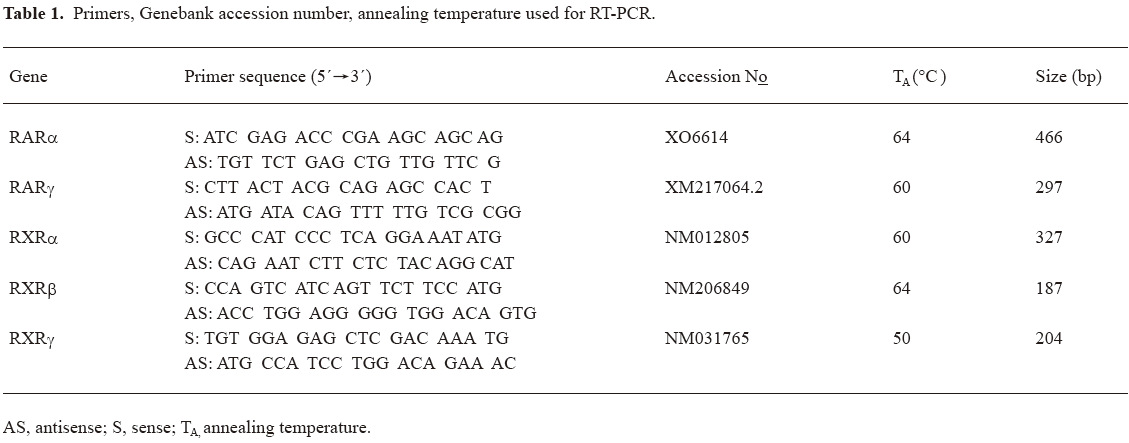
Full table
Western blot analysis The mesangial cell monolayer was washed 3 times with ice-cold phosphate-buffered solution (PBS). Cells and matrix were lysed with 1× reducing Laemmli buffer containing a 1:50 dilution of a protease inhibitor cocktail (Roche) on ice, scraped off the plates. Cell debris was pelleted by centrifugation (10 000×g, 10 min, 4 °C). Supernatant was collected and protein concentrations measured using BCA protein assay (Shenergy Biocolor Co). Equal amounts of protein from samples were loaded and separated under reducing conditions on SDS-PAGE (7.5% w/v for fibronectin and 10% for PAI-1 and β-actin). Prestained molecular-mass standards were used to monitor protein migration. Proteins were transferred onto PVDF membranes (Amersham Bioscience). The membranes were blocked in 5% skim milk in Tris-buffered saline with 0.1% Tween 20 (TBST), then incubated with the primary antibody against fibronectin (Chemicon), PAI-1 (BD Biosciences/Pharmingen) and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by corresponding peroxidase-conjugated secondary antibody. Immune complexes were detected using the enhanced chemiluminescence (ECL) reagents (Pierce); immunoreactive bands were quantified using Smart viewer software (Furi Technology Co, Shanghai, China). The results were normalized against the intensity of the internal control (β-actin) band for each sample.
Statistical analysis Data were expressed as the mean±SD. Statistical analysis was carried out using Sigma stat 2.0 software. Differences in mean values between groups were analyzed using ANOVA followed by Student-Newman-Keuls test or by Dunn multiple comparison test as appropriate. A P value of <0.05 was considered statistically significant.
Results
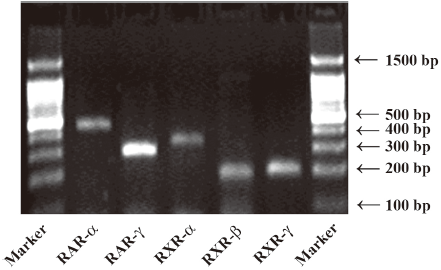
mRNA expression of RA receptors in cultured rat mesangial cells Using RT-PCR analysis, we identified the presence of RAR-α, -γ and RXR-α, -β, -γ mRNA in cultured rat glomerular mesangial cells (Figure 1).
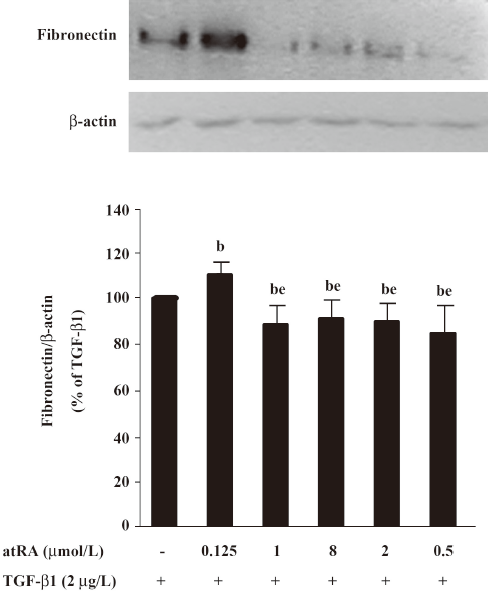
Effect of atRA on fibronectin expression induced by TGF-β1 and Ang II Our preliminary study showed that atRA alone was toxic only at 10 μmol/L, slightly increased the basal fibronectin level at 2 μmol/L, unexpectedly and markedly increased the basal fibronectin level at 0.2 μmol/L or 0.02 μmol/L with lower concentration being more effective (data not shown). Next we used atRA within a range from 0.125 to 8 μmol/L and found that pretreatment with atRA at a concentration of 0.5 to 8 μmol/L attenuated TGF-β1-induced fibronectin expression (P<0.05 vs TGF-β1) showing no dose-dependent difference, whereas a lower atRA concentration (0.125 μmol/L) increased TGF-β1-induced fibronectin expression (P<0.05 vs TGF-β1) (Figure 2). Here we focused on the inhibitory effect of atRA, so the concentrations of 2 and 0.5 μmol/L were chosen in the present study.
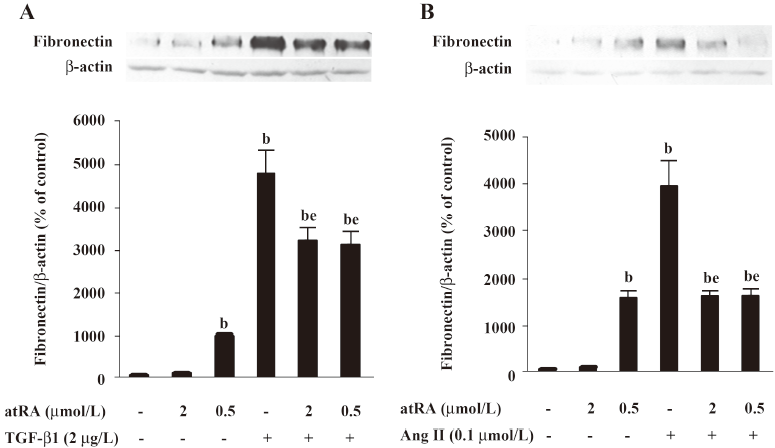
As shown in Figure 3A and 3B, 2 μmol/L atRA alone slightly increased, and 0.5 μmol/L atRA alone markedly increased the basal fibronectin level (P<0.05 vs control). However, pretreatment with either 2 μmol/L or 0.5 μmol/L atRA effectively inhibited the increase in fibronectin induced by TGF-β1 (2 μg/L) and Ang II (0.1 μmol/L) for 48 h in rat mesangial cells (P<0.05 vs TGF-β1 or Ang II) showing no dose-dependent difference.
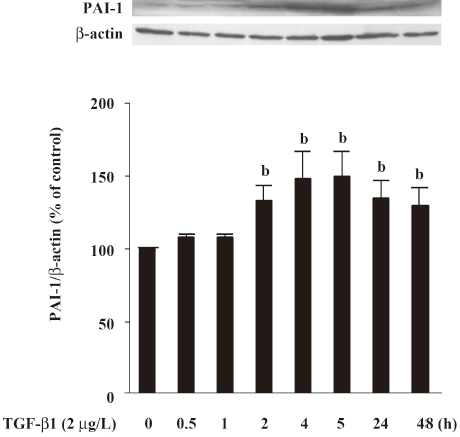
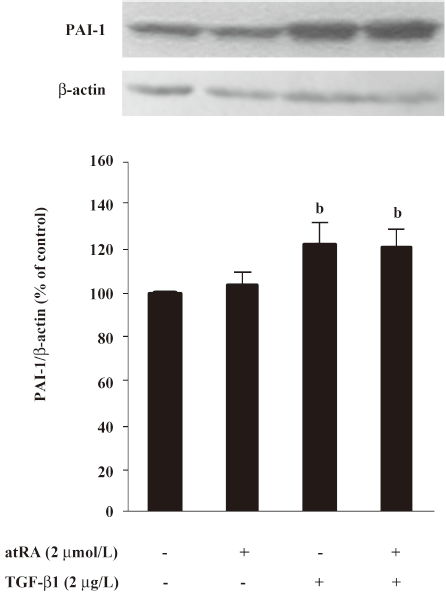
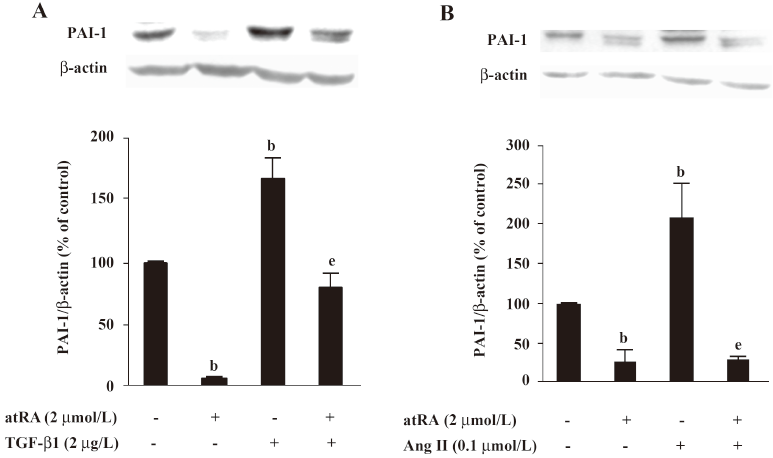
Effect of atRA on PAI-1 expression induced by TGF-b1 and Ang II PAI-1 protein was increased 2 h after TGF-β1 (2 μg/L) stimulation, peaked at 5 h and decreased at 48 h (Figure 4). As illustrated in Figure 5, the early (5 h) basal and early TGF-β1-induced PAI-1 expressions were not affected by atRA. However, the sustained (48 h) PAI-1 expression under both basal and TGF-β1-stimulated conditions was markedly attenuated by atRA (P<0.05) (Figure 6A). The sustained (48 h) PAI-1 expression under both basal and Ang II-stimulated conditions was also markedly diminished by atRA (P<0.05) (Figure 6B).
Discussion
The present study provides the first piece of direct evidence about the inhibitory effect of atRA on fibronectin production in mesangial cells. This effect of atRA is associated with a change in PAI-1 levels.
PAI-1 is the major physiological inhibitor of plasmin generation. High levels of PAI-1 are believed to favor the development of fibrosis, presumably because plasmin degrades ECM and activates other ECM-degrading MMPs. ECM accumulation is aggravated by PAI-1 overexpression suggesting the involvement of PAI-1 in fibrosis[4]. Moreover, fibrosis is ameliorated by PAI-1 depletion or blockage in both in vitro and in vivo experiments[2,5–9]. It is noteworthy that plasmin-independent mechanisms may also be implicated in profibrotic effects of PAI-1, because the de-adhesive properties of PAI-1 do not require plasminogen or the generation of plasmin, and may play a role in promoting fibrosis by recruiting leukocytes and matrix-producing cells into the damaged tissue[22,23]. TGF-β1 and Ang II have been well recognized as major mediators in progressive glomerulosclerosis[10,13]. TGF-β1 stimulates the expression of ECM proteins such as collagens, laminin and fibronectin. On the other hand it suppresses the degradation of ECM by increasing PAI-1 synthesis [10-12]. Ang II also upregulates genes encoding ECM and PAI-1 proteins[13–15]. Its profibrotic action has been shown to be partially mediated by TGF-β1[14,15]. Therefore, we investigated the in vitro effect of atRA on fibronectin and PAI-1 expression induced by TGF-β1 and Ang II to further explore the mechanisms underlying the beneficial action of atRA on renal fibrosis.
In the present study, rat glomerular mesangial cells expressed five of the six RA receptor subtypes mRNA (only RAR-β is absent) using RT-PCR analysis, suggesting that these receptors are present in mesangial cells. The present study showed that TGF-β1 rapidly induced a sustained increase in PAI-1 protein in mesangial cells. Interestingly, atRA did not alter the early (5 h) increase in PAI-1 but markedly inhibited the prolonged (48 h) PAI-1 induction. We assume that the mechanisms for PAI-1 induction at early time points are different from those at late time points. atRA may only interfere with the latter mechanisms. Another possible explanation is that atRA generates another mediator, which in turn exerts antifibrotic effects. Thus the inhibitory effect of atRA on PAI-1 expression occurs relatively late. For example, 9-cis RA, an isomer of atRA, has been reported to induce mRNA and protein expression of hepatocyte growth factor (HGF) at 24 h and 72 h in cultured mesangial cells[24]. It is possible that atRA inhibition of fibronectin expression stimulated by TGF-β1 is mediated by an inhibition of PAI-1 expression. Nevertheless, we cannot exclude that atRA may interfere with TGF-β1-stimulated fibronectin expression at the transcriptional level. In the present study, atRA also inhibited Ang II-induced increase in fibronectin and PAI-1. Previous studies have shown that atRA downregulates Ang II type I receptor mRNA in vascular smooth muscle cells[25] and that Ang II stimulates ECM and PAI-1 synthesis partially through induction of TGF-β1 expression[14,15], so it is expected that fibronectin and PAI-1 induction by Ang II can be repressed by atRA treatment.
atRA decreased fibronectin expression induced by TGF-β1 and Ang II in mesangial cells at concentrations of 2 and 0.5 μmol/L. However, atRA alone increased the basal fibronectin expression in mesangial cells, suggesting that atRA may increase or decrease fibronectin expression depending on the status of the cells in culture, stimulatory or quiescent. In line with our study, Chen et al[26] have reported that atRA alone activates mitogenesis of vascular smooth muscle cells under quiescent conditions. However, atRA inhibits mitogenesis in the presence of endothelin by different signaling pathways. Our data also showed that atRA alone was more effective in increasing the basal fibronectin expression at a very low concentration of 0.02 μmol/L than that at a higher concentration of 0.2 μmol/L. In agreement with our results, atRA alone has been shown to stimulate mitogenesis of vascular smooth muscle cells at low concentrations where the stimulatory effect of atRA at lower concentration is more evident than that at higher concentration[26]. In contrast with the inhibitory effects of atRA at high concentrations (0.5 to 8 μmol/L), it enhanced TGF-β1-induced fibronectin expression at a low concentration of 0.125 μmol/L in mesangial cells. The data suggest that atRA may inhibit or enhance TGF-β1-induced fibronectin expression depending on the concentrations of atRA used. The reasons for this biphasic effect are unclear. However, this is not an unusual biological response. For example, glibenclamide, a stimulator of glucose uptake, exerts opposing effects on high glucose-induced ECM formation at low (0.01 μmol/L) and high (1 μmol/L) concentrations[27]. The mechanisms under this phenomenon remain to be investigated.
At concentrations of 2 and 0.5 μmol/L, atRA was almost equally effective in reducing fibronectin expression stimulated by TGF-β1 and Ang II. A previous study from our laboratory has also shown that the dose-dependence of atRA action on cardiac fibrosis in spontaneously hypertensive rats is not apparent[21]. In addition, we can not show which RA receptor subtype is involved in mediating these effects of atRA. Experimental approaches using mesangial cells deficient in specific RA receptors or synthetic RA receptor ligands may help to address this question.
In summary, the present study shows that atRA at high concentrations inhibits the increase in fibronectin expression induced by TGF-β1 and Ang II in glomerular mesangial cells. This activity of atRA is associated with a change in PAI-1 levels. atRA might be an useful antifibrotic drug in the treatment of kidney diseases.
References
- Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med 1998;339:1448-56.
- Baricos WH, Cortez SL, el-Dahr SS, Schnaper HW. ECM degradation by cultured human mesangial cells is mediated by a PA/plasmin/MMP-2 cascade. Kidney Int 1995;47:1039-47.
- Eddy AA. Plasminogen activator inhibitor-1 and the kidney. Am J Physiol Renal Physiol 2002;283:F209-20.
- Matsuo S, López-Guisa JM, Cai X, Okamura DM, Alpers CE, Bumgarner RE, et al. Multifunctionality of PAI-1 in fibrogenesis: evidence from obstructive nephropathy in PAI-1-overexpressing mice. Kidney Int 2005;67:2221-38.
- Oda T, Jung YO, Kim HS, Cai X, López-Guisa JM, Ikeda Y, et al. PAI-1 deficiency attenuates the fibrogenic response to ureteral obstruction. Kidney Int 2001;60:587-96.
- Nicholas SB, Aguiniga E, Ren Y, Kim J, Wong J, Govindarajan N, et al. Plasminogen activator inhibitor-1 deficiency retards diabetic nephropathy. Kidney Int 2005;67:1297-307.
- Krag S, Danielsen CC, Carmeliet P, Nyengaard J, Wogensen L. Plasminogen activator inhibitor-1 gene deficiency attenuates TGF-beta1-induced kidney disease. Kidney Int 2005;68:2651-66.
- Huang Y, Haraguchi M, Lawrence DA, Border WA, Yu L, Noble NA. A mutant, noninhibitory plasminogen activator inhibitor type 1 decreases matrix accumulation in experimental glomerulonephritis. J Clin Invest 2003;112:379-88.
- Huang Y, Border WA, Lawrence DA, Noble NA. Noninhibitory PAI-1 enhances plasmin-mediated matrix degradation both in vitro and in experimental nephritis. Kidney Int 2006;70:515-22.
- Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC. TGF-beta signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol 2003;284:F243-52.
- Baricos WH, Cortez SL, Deboisblanc M, Xin S. Transforming growth factor-beta is a potent inhibitor of extracellular matrix degradation by cultured human mesangial cells. J Am Soc Nephrol 1999;10:790-5.
- Wilson HM, Reid FJ, Brown PA, Power DA, Haites NE, Booth NA. Effect of transforming growth factor-beta 1 on plasminogen activators and plasminogen activator inhibitor-1 in renal glomerular cells. Exp Nephrol 1993;1:343-50.
- Mezzano SA, Ruiz-Ortega M, Egido J. Angiotensin II and renal fibrosis. Hypertension 2001;38:635-8.
- Kagami S, Kuhara T, Okada K, Kuroda Y, Border WA, Noble NA. Dual effects of angiotensin II on the plasminogen/plasmin system in rat mesangial cells. Kidney Int 1997;51:664-71.
- Kagami S, Border WA, Miller DE, Noble NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-beta expression in rat glomerular mesangial cells. J Clin Invest 1994;93:2431-7.
- Han SY, So GA, Jee YH, Han KH, Kang YS, Kim HK, et al. Effect of retinoic acid in experimental diabetic nephropathy. Immunol Cell Biol 2004;82:568-76.
- Oseto S, Moriyama T, Kawada N, Nagatoya K, Takeji M, Ando A, et al. Therapeutic effect of all-trans retinoic acid on rats with anti-GBM antibody glomerulonephritis. Kidney Int 2003;64:1241-52.
- Schaier M, Liebler S, Schade K, Shimizu F, Kawachi H, Grone HJ, et al. Retinoic acid receptor alpha and retinoid X receptor specific agonists reduce renal injury in established chronic glomerulonephritis of the rat. J Mol Med 2004;82:116-25.
- Lehrke I, Schaier M, Schade K, Morath C, Waldherr R, Ritz E, et al. Retinoid receptor-specific agonists alleviate experimental glomerulonephritis. Am J Physiol Renal Physiol 2002;282:F741-51.
- Wagner J, Dechow C, Morath C, Lehrke I, Amann K, Waldherr R, et al. Retinoic acid reduces glomerular injury in a rat model of glomerular damage. J Am Soc Nephrol 2000;11:1479-87.
- Lü L, Yao T, Zhu YZ, Huang GY, Cao YX, Zhu YC. Chronic all-trans retinoic acid treatment prevents medial thickening of intramyocardial and intrarenal arteries in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 2003;285:H1370-7.
- Czekay RP, Loskutoff DJ. Unexpected role of plasminogen activator inhibitor 1 in cell adhesion and detachment. Exp Biol Med (Maywood) 2004;229:1090-6.
- Stefansson S, Lawrence DA. The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature 1996;383:441-3.
- Wen X, Li Y, Hu K, Dai C, Liu Y. Hepatocyte growth factor receptor signaling mediates the anti-fibrotic action of 9-cis-retinoic acid in glomerular mesangial cells. Am J Pathol 2005;167:947-57.
- Haxsen V, Adam-Stitah S, Ritz E, Wagner J. Retinoids inhibit the actions of angiotensin II on vascular smooth muscle cells. Circ Res 2001;88:637-44.
- Chen S, Gardner DG. Retinoic acid uses divergent mechanisms to activate or suppress mitogenesis in rat aortic smooth muscle cells. J Clin Invest 1998;102:653-62.
- Giannico G, Cortes P, Baccora MH, Hassett C, Taube DW, Yee J. Glibenclamide prevents increased extracellular matrix formation induced by high glucose concentration in mesangial cells. Am J Physiol Renal Physiol 2007;292:F57-65.