
5-Hydroxydecanoate inhibits proliferation of hypoxic human pulmonary artery smooth muscle cells by blocking mitochondrial KATP channels1
Introduction
Constriction and remodeling of the pulmonary arteries induced by hypoxia are very important physiological phenomena in order to maintain ventilation–perfusion matching. However, a sustained hypoxic pulmonary vasoconstriction and artery remodeling also contribute to hypoxic pulmonary hypertension. Until now, the mechanism of hypoxic pulmonary hypertension has still not been fully elucidated. The proliferation of pulmonary artery smooth muscle cells plays an important role in hypoxic pulmonary artery remodeling. The imbalance between apoptosis and proliferation may contribute to this cell proliferation[1,2]. Mitochondria play a great part in regulating cell apoptosis and proliferation[3]. When mitochondrial membrane potential (Δψm) collapses, cytochrome c, which normally exists in the mitochondrial intermembrane space, is released from the mitochondrial intermembrane space to the cytosol. An increase in cytosolic cytochrome c is a trigger for the activation of caspase-9, which then activates a set of cysteine proteases that are central executioners of the cell apoptotic pathway. The mitochondrial membrane ATP-sensitive potassium channel (MitoKATP) is a key factor which determinesΔψm[4]. MitoKATP is very sensitive to hypoxia. In a normoxic environment, MitoKATP always is closed and Δψm collapses; while in a hypoxic environment, MitoKATP is opened and Δψm increases. Recently, some studies have demonstrated that MitoKATP contributes to apoptosis and proliferation of myocardial cells[5,6]. So far, there has not been any reports as to whether MitoKATP plays any important role in human hypoxic pulmonary artery remodeling. Diazoxide, an opener of MitoKATP, and 5-hydroxydecanoate (5-HD), an antagonist of MitoKATP, were used to influence MitoKATP. Both diazoxide and 5-HD, which are specific for KATP on the mitochondrial membrane, have no effect on cell membrance. This study was aimed to investigate the roles of MitoKATP and Δψm in the proliferation of hypoxic human pulmonary artery smooth muscle cells and the pharmacological effect of 5-HD on this process.
Materials and methods
Ethics of experimentation This study was approved by the ethical committee in our hospital and by the patients involved in this study. The experiments were conducted in accordance with the Declaration of Helsinki.
Culture and identification of human pulmonary artery smooth muscle cells Normal human lung tissues were obtained from 6 patients receiving partial lung resection. Pulmonary arteries (diameter: 2–3 mm) were dissected from the surrounding parenchyma and washed twice in precooled D-Hanks’ solution containing 100 U/mL penicillin and 100 U/mL streptomycin. Under a dissecting microscope, smooth muscle bundles were dissected from the artery. The minced muscle fragments (about 1 mm3 in size) were digest-ed in D-Hanks’ solution containing 3 mg/mL collagenase type I (Sigma, St Louis, MO, USA) at 37 °C for 50–60 min. Then 1–2 mL of 0.25% trypsin (Sigma, St Louis, MO, USA) was added and incubated for about 10 min for digestion. The digestion was finally stopped with Dulbecco’s modified Eagle’s medium (DMEM) containing 20% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA). The dissociated cells were resuspended in DMEM containing 20% FBS, 100 U/mL penicillin, and 100 U/mL streptomycin, seeded onto 50 mL culture flasks and maintained at 37 °C in an humidified atmosphere with 5% CO2. The culture medium was replaced every 2–3 d until conflu-ence was achieved (at d 15–20). The cells were digested with 0.25% trypsin and subcultured following the same procedure. Smooth muscle cells were confirmed by immunohistochemistry staining against α-actin using a mouse antihuman smooth muscle α-actin polyclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and by the typical “hill and valley” morphological pattern of confluent smooth muscle cells under phase contrast microscope.
Grouping of human pulmonary artery smooth muscle cells HPASMC (passages 4–8) were cultured in DMEM with 20% FBS and divided into 6 groups: (i) control group, where cells were cultured in normoxia for 24 h (20% O2, 5% CO2); (ii) diazoxide group, where cells were pretreated with a MitoKATP specific opener-diazoxide[8] (100 µmol/L; Sigma, USA) and cultured in normoxia for 24 h. Diazoxide was dissolved in dimethylsulfoxide(DMSO) before being added to the experimental solution (DMEM without FBS). The final concentration of DMSO in DMEM was <0.1%[8]; (iii) 5-HD group, where cells were pretreated with a MitoKATP specific antagonist–5-HD [8] (500 μmol/L Sigma, USA) and cultured in normoxia for 24 h. 5-HD was dissolved in the experimental solution (DMEM without FBS); (iv) 24 h hypoxia group, where cells were cultured at 37 °C in an atmosphere containing 5% O2, 5% CO2, and 90% N2 for 24 h; (v) 24 h hypoxia+diazoxide group, which involved 24 h hypoxia treatment in the concomitant stimulation of diazoxide; and (vi) 24 h hypoxia+5-HD group, which involved 24 h hypoxia treatment in the concomitant stimulation of 5-HD.
Measurement of rhodamine-123 fluorescence[9] The cells seeded on 25 mm coverslips were stained with rhodamine 123 (R-123; Sigma, St Louis, MO, USA) by incubating with 10 µg/mL R-123 for 30 min at 37 °C. The cells were perfused with physiological salt solution to establish a baseline of fluorescence. R-123 fluorescence was excited at 488 nm and measured at 530 nm with Leica SP-1 confocal microscopy (Leica, wiesbaden, Germany). The fluorescence signals were stored in a computer and analyzed with Leica TCS NT software.
R-123 is taken up selectively by mitochondria[10,11] and its uptake depends on Δψm. In isolated mitochondria, the relationship between the intensity of R-123 fluorescence and Δψm is linear. The uptake of R-123 fluorescence, which is quenched at resting Δψm, increases with the depolarization of the mitochondrial membrane[11].
Measurement of HPASMC proliferation by 3-(4,5-dimethyl-2-thiazol-yl)-2,5- diphenyl-2H-tetrazolium bromide (MTT) assay HPASMC were seeded at a density of 1×104 cells/well into 96-well plates, cultivated, and divided into different groups as described above. MTT (5 mg/mL) was added to the wells (20 µL/well) at the end of the experimental period. After 4 h incubation at 37 °C, media were removed from the wells, and DMSO was added (150 µL/well). The plates were agitated at room temperature for 30 min. Absorbance (value A) of every well at a 490 nm wavelength was read on an ELISA reader.
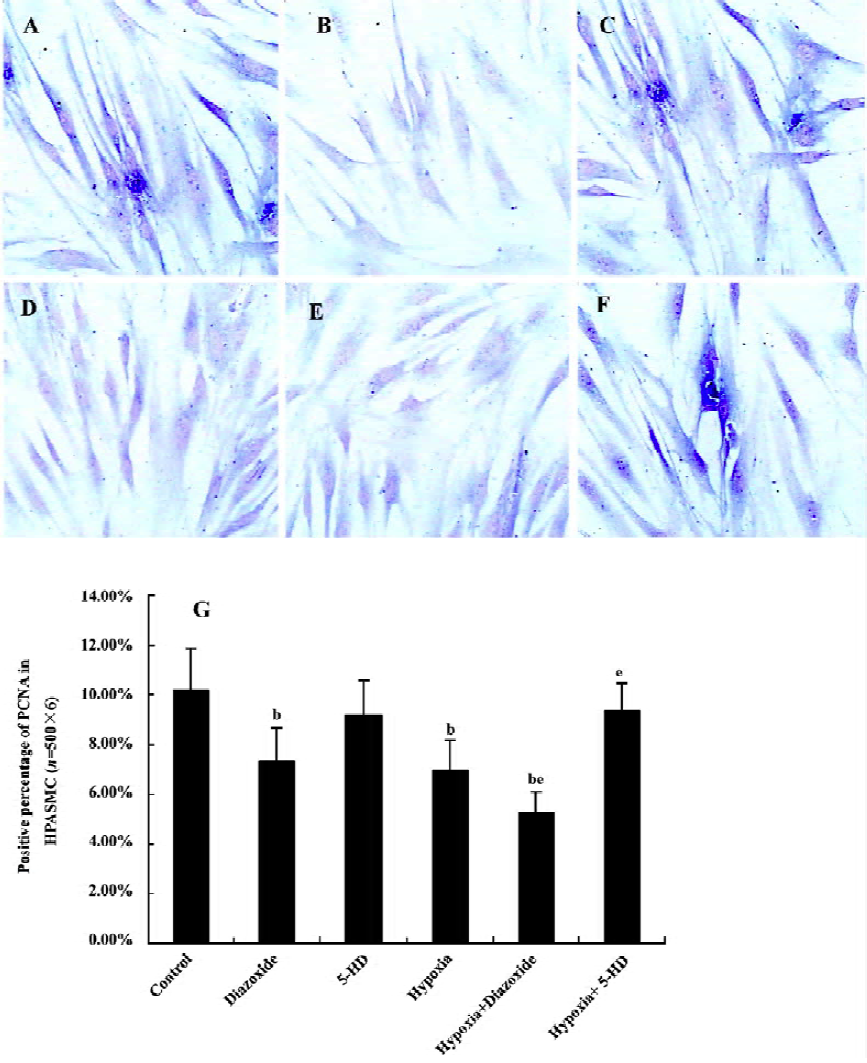
Detection of expression of proliferative cell nuclear antigen in HPASMC by immunohistochemistry staining HPASMC were seeded at a density of 1×105 cells/mL onto 6-well plates, cultivated, and divided into the 6 different groups as described earlier. The HPASMC were fixed with cool acetone for 10 min. The expression of proliferative cell nuclear antigen (PCNA) in the HPASMC was detected by immunohistochemistry staining using a polyclonal antibody against PCNA (Santa Cruz, USA) at 4 °C overnight. The same process without the primary antibody was used as a control. After incubation with the secondary antibody (Beijing Zhongshan Biotechnology, Beijing, China) at 37 °C for 1 h, the HPASMC were observed under light microscopy and recorded photographically. The criteria for judgement were as follows: the cells with brown staining in nuclei were PCNA positive. On each slide, 500 cells (with the detection of 6 slides) were randomly counted under a light microscope and then the positive percentage per 100 cells was calculated.
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling technique For the detection of apoptotic cells, the apoptotic index was examined by the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling (TUNEL) method. An in situ cell death detection kit (ISCDD, Boehringer Mannheim, Germany) was used to detect the apoptotic cells. The procedures were carried out according to the protocol of the kit and other references. The HPASMC were observed under microscopy at once and recorded photographically. The cells with deep blue staining in nuclei were TUNEL positive. In each slide (6 slides), 500 cells were randomly counted under a light microscope and then the positive percentage per 100 cells was calculated.
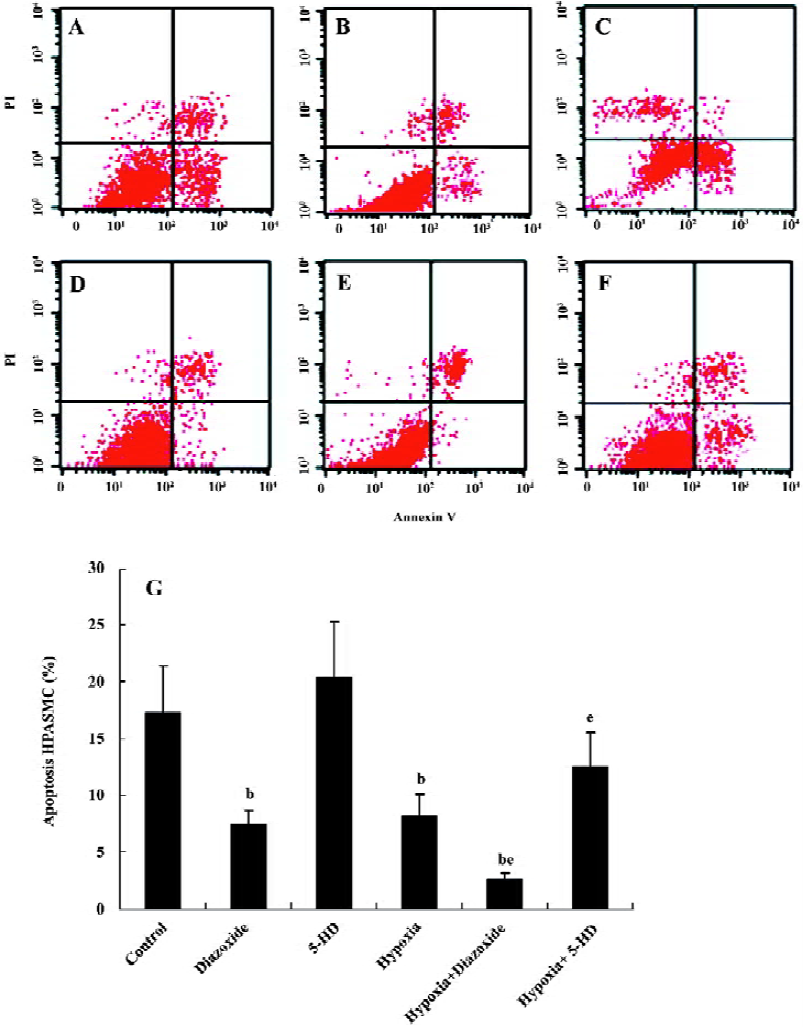
Flow cytometry analysis To determine the apoptosis, the treated cells (1×109 cells/L) were washed in phosphate-buffered saline (PBS) and fixed in 70% ethanol at 4 °C for 12 h. After incubation, 195 µL of the solution was transferred to a 5 mL culture tube with 5 µL Annexin V-fluorescein isothio-cyanate (FITC) added. The tube was then incubated for 30 min at room temperature in the dark. The cells were washed with PBS and resuspended in 190 µL binding buffer with 10 µL propidium iodide (PI) added. Finally, the tube was gently vortexed and incubated for another 30 min in the dark and then the cells were analyzed immediately by flow cytometry. Samples were acquired on a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, New Jersey, USA) and analyzed with CellQuest software (Becton Dickinson, Franklin Lakes, New Jersey, USA). Early apoptotic cells were characterized by high Annexin binding and low PI staining, whereas late apoptotic and necrotic cells were stained strongly with both Annexin and PI.
Statistical analysis All data were expressed as mean±SD. ANOVA was used for the comparison of variance among several groups. The significance of difference between 2 groups was tested by t-test. P<0.05 was considered statistically significant.
Results
Cell identification As shown in Figure 1, under inverted phase contrast microscope, the HPASMC showed spindle-shaped features, central oval nuclei with prominent nucleoli, as well as the characteristic “hill and valley” appearance. Immunohistochemistry staining of smooth muscle-specific α-actin was positive. These results indicated that the cells in culture are smooth muscle cells.
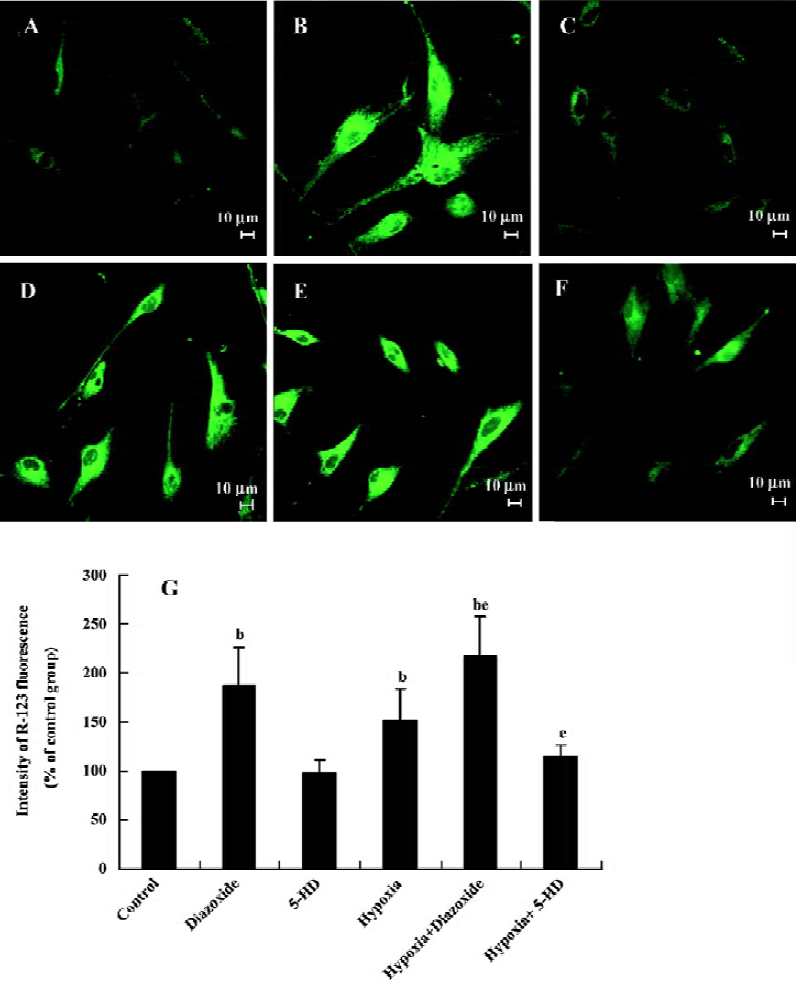
Changes of HPASMC mitochondrial membrane potential in HPASMC As shown in Figure 2, diazoxide (100 µmol/L), a MitoKATP specific opener, increased the intensity of R-123 fluorescence in the HPASMC as compared with the control group (increased by 87%±39.86%, n=6, P<0.05; Figure 2), indicating an increase of mitochondrial membrane potential. However, 5-HD (500 µmol/L) did not markedly change the intensity of R-123 fluorescence in the HPASMC (n=6, P>0.05; Figure 2). Both 24 h hypoxia treatment and 24 h hypoxia treatment in the concomitant stimulation of diazoxide increased R-123 fluorescent intensity (increased by 52%±32.42%, and 117%±40.17%, respectively; Figure 2). The changes of R-123 fluorescence intensity were obviously enhanced under 24 h hypoxia treatment in the concomitant stimulation of diazoxide as compared to 24 h hypoxia treatment or diazoxide stimulation alone (217%±40.17% vs 152%±32.42% and 217%±40.17% vs 187%±39.86%, respec-tively, n=6, P<0.05; Figure 2). 5-HD attenuated a 24 h hypoxia-induced increase in R-123 fluorescence intensity (114%±12.57% vs 152%±32.42%, n=6, P<0.05; Figure 2). These results imply that hypoxia opens MitoKATP and induces an increase in Δψm in HPASMC.
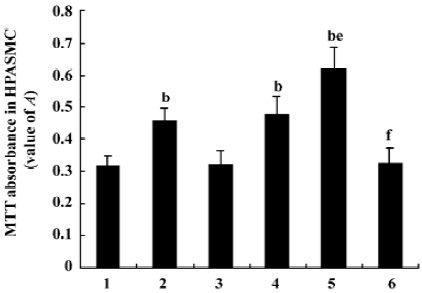
Measurement of HPASMC proliferation by MTT colorimetric assay Diazoxide increased the value A of HPASMC as compared with the control group (0.457%±0.041 vs 0.318%± 0.032%, n=6, P<0.05; Figure 3). However, 5-HD did not markedly change the value A of HPASMC compared with the control group (0.323%±0.039% vs 0.318%±0.032%, n=6, P>0.05; Figure 3). Both 24 h hypoxia treatment and 24 h hypoxia treatment in the concomitant stimulation of diazoxide increased the value A of HPASMC (0.478%±0.058% vs 0.318%±0.032% and 0.621%±0.067% vs 0.318%±0.032%, respectively, n=6, P<0.05; Figure 3). Diazoxide significantly enhanced the effects of 24 h hypoxia on the value A of HPASMC (0.621%±0.067% vs 0.478%±0.058%, n=6, P<0.05; Figure 3). 5-HD could weaken the increasing effect of 24 h hypoxia on the value A of HPASMC (0.328%±0.044% vs 0.478%±0.058%, n=6, P<0.05; Figure 3). These results demonstrate that hypoxia and diazoxide induced cell proliferation; 5-HD almost completely inhibited the increase of value A induced by hypoxia treatment.
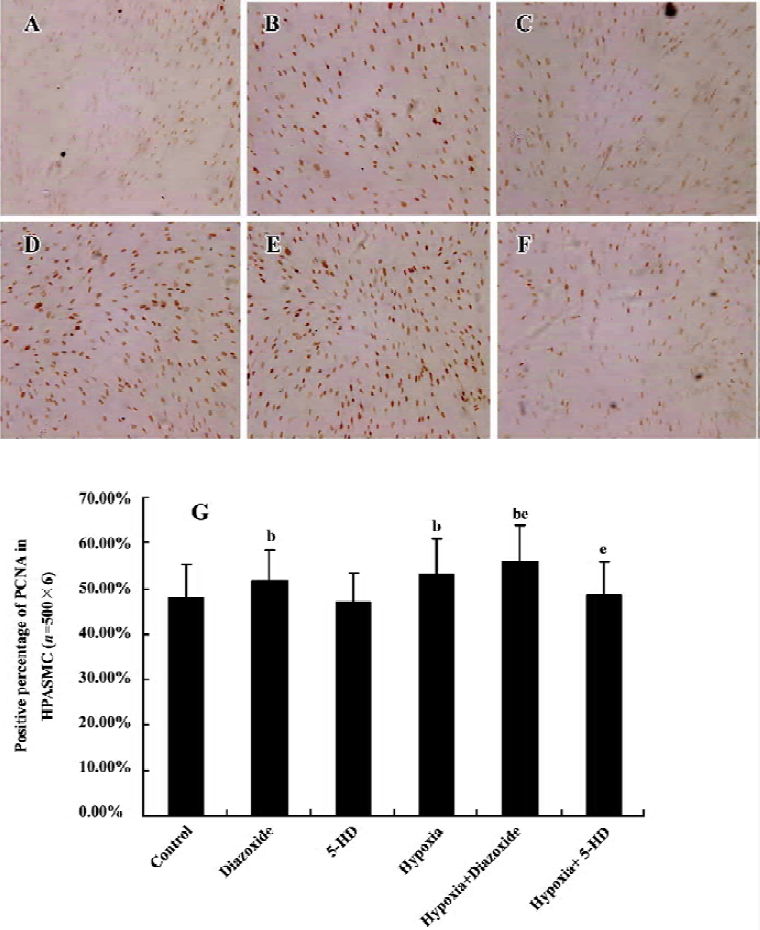
Expression of PCNA in HPASMC Diazoxide increased the expression of PCNA compared with the control group (P<0.05; Figure 4). However, 5-HD did not markedly change the expression of PCNA compared with the control group (n=6, P>0.05; Figure 4). The 24 h exposure of the HPASMC to hypoxia increased the PCNA expression (n=6, P<0.05; Figure 4) and this was significantly augmented by a concomitant stimulation of diazoxide (n=6, P<0.05; Figure 4). 5-HD obviously attenuated a 24 h hypoxia-increased expression of PCNA (n=6, P<0.05; Figure 4). These results demonstrated that both hypoxia and diazoxide induced the proliferation of the HPASMC, a process that can be inhibited by 5-HD.
TUNEL staining Diazoxide decreased the rate of positive TUNEL-staining cells in the HPASMC compared with the control group (n=6, P<0.05; Figure 5). However, 5-HD did not markedly alter the TUNEL staining result in the HPASMC compared with the control group (n=6, P>0.05; Figure 5). The exposure of the HPASMC to hypoxia for 24 h prominently decreased the rate of positive TUNEL-staining cells (n=6, P<0.05; Figure 5).These effects of hypoxia on TUNEL staining is obviously augmented by the concomitant treatment of diazoxide (n=6, P<0.05; Figure 5), whereas 5-HD significantly reversed the hypoxia-induced decline in TUNEL staining in the HPASMC (n=6, P<0.05; Figure 5). These results demonstrate that both hypoxia and diazoxide can inhibit apoptosis in HPASMC, which can be reversed by 5-HD.
Flow cytometric analysis To determine whether 5-HD and diazoxide affect the apoptotic rate in HPASMC, we also examined the expression of Annexin V and the exclusion of PI by flow cytometry analysis. Diazoxide decreased the percentage of apoptosis in HPASMC compared with the control group (7.41%±1.32% vs 17.23%±4.16%, n=6, P<0.05; Figure 6). However, 5-HD did not markedly alter the positive percentage of apoptosis compared with the control group (20.37%±4.89% vs 17.23%±4.16%, n=6, P>0.05). The exposure of HPASMC to hypoxia for 24 h prominently decreased the positive percentage of apoptosis (8.27%±1.82% vs 17.23%± 4.16%, n=6, P<0.05). These effects of hypoxia on the positive percentage of apoptosis are obviously decreased by the concomitant treatment of diazoxide (2.52%±0.22% vs 8.27%±1.82%, n=6, P<0.05), whereas 5-HD significantly reversed the hypoxia-induced decline in the positive percentage of apoptosis in HPASMC (12.48%±3.07% vs 8.27%±1.82%, n=6, P<0.05). These results also demonstrate that both hypoxia and diazoxide inhibit apoptosis in HPASMC, which can be reversed by 5-HD.
Discussion
The precise control of the balance between pulmonary artery smooth muscle cell (PASMC) proliferation and apoptosis plays an important role in maintaining normal pulmonary vascular structure and function. The reinforcement of proliferation or the weakness of apoptosis in PASMC leads to pulmonary hypertension and pulmonary vascular structure remodeling. Apoptosis is a physiological mode of cell death that is triggered by diverse external or internal signals. Apoptosis in pulmonary artery smooth muscle cells allows the pulmonary vasculature to tightly control the vascular wall tissue mass (or thickness). Dysfunction of this process has been widely linked to the pathogenesis of pulmonary vascular diseases[12]. Some data have demonstrated that apoptosis in PASMC and endothelial cells is involved in the regression of pulmonary arterial hypertrophy[13]. Most pulmonary hypertension was caused by hypoxia induced by various reasons[14]; this pulmonary hypertension was named hypoxic pulmonary hypertension. Some studies have shown that hypoxia can promote PASMC proliferation, which can subsequently trigger hypoxic pulmonary vascular structure remodeling[14]. The mechanism of hypoxic pulmonary vascular structure remodeling has not yet been fully elucidated, and studies about the mechanism on human PASMC are very scarce. It is a complex network that relates to many cytokines and transcriptional factors.
Mitochondrial is a very important oxygen sensor in mammalian cells. However, the studies about the modulation mechanism of mitochondrial on hypoxic pulmonary vascular structure remodeling are rarely documented. Recently, some studies have found that Δψm was markedly linked to cell proliferation in myocardial cells[15], but studies about Δψm are seldom on PASMC. It is known that MitoKATP is extremely sensitive to hypoxia[16] and is a major factor in the control of Δψm. Furthermore, some studies have identified a mechanistic link between MitoKATP channels and the mitochondrial apoptotic pathway. The principal findings are as follows: (i) in isolated cardiac myocytes, the MitoKATP channel opener diazoxide inhibits the activation of the mitochondrial apoptotic pathway induced by oxidative stress in a concentration-dependent manner[17]; And (ii) the channel blocker 5-HD (as well as glibenclamide) abolishes the anti-apoptotic effect of diazoxide[18]. These observations support the hypothesis that the activation of MitoKATP channels inhibits apoptosis[19]. In a normoxic condition, the production of ATP is at a natural level and the MitoKATP is always closed. Mean-while, Δψm collapses and cytochrome c is released from the mitochondrial intermembrane space to the cytosol. An increase in cytosolic cytochrome c is a trigger for the activation of caspase-9, which then activates a set of cysteine proteases that are central executioners of the cell apoptotic pathway[20]. In a hypoxic environment, the production of ATP decreases[21], which triggers the activation of MitoKATP and the Δψm increase, subsequently inhibits cytochrome c releasing from the mitochondrial intermembrane space to the cytosol and triggers cell proliferation. So far, it has not been reported whether MitoKATP plays an important role in human hypoxic pulmonary vascular structure remodeling.
In the present study, hypoxia for 24 h increased the value of MTT and PCNA staining, decreased TUNEL staining, as well as the percentage of apoptotic cells in HPASMC. These results showed that hypoxia promoted HPASMC proliferation and inhibited HPASMC apoptosis. Diazoxide opened MitoKATP, increased Δψm and the proliferative status of HPASMC in a similar way as that of HPASMC exposed to hypoxia. These results also showed that an increase in Δψm promoted HPASMC proliferation. In the presence of 5-HD, MitoKATP was closed and the proliferative status of HPASMC showed no any remarkable change compared with the control group. This study has shown that an exposure to hypoxia for 24 h increases Δψm and promotes HPASMC proliferation. 5-HD can inhibit MitoKATP and therefore attenuate hypoxia- and diazoxide-induced proliferation in HPASMC. These results prove that MitoKATP plays an important role in HPASMC proliferation induced by hypoxia. In addition, many other factors may be involved in this signaling pathway, for instance, hypoxia-inducible factor 1 [22], protein kinase C[23], fas/fasL[23], c-jun, Bcl-2[24], [Ca2+]i[25]. Other factors that might be involved in the mechanism of hypoxic pulmonary vascular structure remodeling remain to be investigated.
In the present study, we found that 5-HD can prevent hypoxic pulmonary artery smooth muscle cell proliferation. We speculate that the mechanisms of 5-HD on hypoxic pulmonary vascular structure remodeling may involve several routes as follows: (i) to change mitochondrial morphology and structure via changing mitochondrial membrane potential; (ii) to change the transmission and metabolism of cell energy; (iii) to impact other cellular factors involved in pulmonary artery smooth muscle cell proliferation; and (iv) other potential routes. In future, we will keep focusing on the effects of 5-HD in vivo (animal model) and further investigate the pharmacological mechanisms of 5-HD as an inhibitor of mitochondrial KATP channel activation on hypoxic pulmonary vascular structure remodeling.
In summary, our study suggests that MitoKATP and Δψm play an important role in hypoxic pulmonary vascular structure remodeling in humans, providing theoretical evidence for the further study of the mechanism and prevention of human pulmonary hypertension.
Acknowledgement
We thank all of the surgeons in the Department of Thoracic Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology for their assistance.
References
- Zhang HP, Xu YJ, Zhang ZX, Ni W, Chen SX. Effect of protein kinase C-nuclear factor-kappa B signal transduction pathway on proliferation and expression of vascular endothelial growth factor in human pulmonary artery smooth muscle cells. Zhonghua Jie He He Hu Xi Za Zhi 2004;27:218-23.
- Zhang S, Fantozzi I, Tigno DD, Yi ES, Platoshyn O, Thistlethwaite PA, et al. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2003;285:L740-54.
- Nicholas J, Santos A, Eric D. Essential role of mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001;29:549-54.
- Lobella S, Martha M, Salih S. Mitochondrial KATP channel openers activate the ERK kinase by an oxidant-dependent mechanism. Am J Physiol 2002;283:C273-281.
- O’Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res 2004;94:420-32.
- Zhu YH, Wang X. Overexpression of heat-shock protein 20 in rat heart myogenic cells confers protection against simulated ischemia/reperfusion injury. Acta Pharmacol Sin 2005;26:1076-80.
- Ardehali H. Role of the mitochondrial ATP-sensitive K(+) channels in cardioprotection. Acta Biochim Pol 2004;51:379-90.
- Murata M, Akao M, O'Rourke B, Marban E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca2+ overload during simulated ischemia and reperfusion. Circ Res 2001;89:891-8.
- Stefanie K, Oleksandr P. Augmented K+ currents and mitochondrial membrane depolarization in pulmonary artery myocyte apoptosis. Am J Physiol Lung Cell Mol Physiol 2001;281:L887-94.
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostasis sensor to calcium signalling and cell death. J Physiol (Lond) 1999;516:1-17.
- Duchen MR, Biscoe TJ. Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. J Physiol (Lond) 1992;450:33-61.
- Wang YF, Tian H, Tang CS, Jin HF, Du JB. Nitric oxide modulates hypoxic pulmonary smooth muscle cell proliferation and apoptosis by regulating carbon monoxide pathway. Acta Pharmacol Sin 2007;28:28-35.
- Taraseviciene-Stewart L, Scerbavicius R, Choe KH, Cool C, Wood K, Tuder RM, et al. Simvastatin causes endothelial cell apoptosis and attenuates severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2006;291:L668-76.
- Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 2006;99:675-91.
- Sack MN. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc Res 2006;72:210-9.
- Wang T, Zhang ZX, Xu YJ. Effect of mitochondrial KATP channel on voltage-gated K+ channel in 24 hour-hypoxic human pulmonary artery smooth muscle cells. Chin Med J (Engl) 2005;118:12-9.
- Eigel BN, Gursahani H, Hadley RW. ROS are required for rapid reactivation of Na+/Ca2+ exchanger in hypoxic reoxygenated guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol 2004;286:H955-63.
- Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 2004;287:H841-9.
- Akao M, Ohler A, O’Rourke B, Marbán E. Mitochondria ATP-sensitive potassium channels inhibit apoptosis induced by oxidative stress in cardiac cells. Circ Res 2001;88:1267-75.
- Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Cytochrome c activates K+ channels before inducing apoptosis. Am J Physiol Cell Physiol 2002;283:C1298-305.
- Quayle JM, Turner MR, Burrell HE, Kamishima T. Effects of hypoxia, anoxia, and metabolic inhibitors on KATP channels in rat femoral artery myocytes. Am J Physiol Heart Circ Physiol 2006;291:H71-80.
- Rose F, Grimminger F, Appel J, Heller M, Pies V, Weissmann N, et al. Hypoxic pulmonary artery fibroblasts trigger proliferation of vascular smooth muscle cells: role of hypoxia-inducible transcription factors. FASEB J 2002;16:1660-1.
- Mireia GA, Carl D, John A. Protein kinase C (PKC) inhibits Fas receptor induced apoptosis through modulation of the loss of K and cell shrinkage. J Biol Chem 2000; 30: 19 609–19.
- Daryoush E, Oleksandr P, Stefanie K. Bcl-2 decreases voltage-gated K+ channel activity and enhances survival in vascular smooth muscle cells. Am J Physiol Cell Physiol 2001;281:C157-65.
- Lipskaia L, Lompre AM. Alteration in temporal kinetics of Ca2+ signaling and control of growth and proliferation. Biol Cell 2004;96:55-68.