
Roles of PI3-K/Akt pathways in nanoparticle realgar powders-induced apoptosis in U937 cells
Introduction
Realgar, an arsenic disulfide compound, has long been used as a therapeutic agent to treat some diseases in ancient China and Europe, and its medicinal effects have attracted increasing attention in recent years. It was found that realgar was usually in the treatment of leukemia in China. Previous studies showed that realgar could inhibit proliferation and induce differentiation in the human promyelocytic leukemia cell line HL-60 through activating some serine/threonine protein phosphatases[1]. Although recent evidence has suggested that in vitro realgar exhibits significant antiproliferation effects on many kinds of leukemia, such as HL-60 and K562[2,3], its mechanism of action in human histocytic lymphoma U937 is unclear. Our previous study has shown that in nanoparticle realgar powder (NRP)-induced U937 cell death, the ratio of the Bax/Bcl-2 protein expression was increased. Additionally, the simultaneous activation of c-Jun NH2-terminal kinase (JNK), but not p38 or the extracellular regulated kinase (ERK), was involved in the NRP-induced U937 apoptosis. In the present study, we further investigated the mechanism involved in the NRP-induced U937 cell death.
Apoptosis, or programmed cell death, is a genetically regulated self-destructive cellular death process that is important in development, tissue remodeling, immune regulation, and many diseases[4–7]. Aberrant apoptosis is the major cause for tumor development and progression. The development of cancer is associated with several key events, including deregulated cell growth in response to oncogene activation that resists cells to apoptosis[8,9]. It is well known that among the proteins involved in the regulation of cell death, p53 and p53-regulated proteins, the mitochondrial pathway-related protein a nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase (SIRT1) and the phosphoinositide 3-kinase (PI3-K)/Akt pathways are the key factors in promoting or inhibiting apoptosis.
As a tumor suppressor, p53 enforces normal growth control and genomic stability. A direct inactivation of the gene that encodes it is reflected in the fact that acquired mutations in it or its upstream activators are found in all major human cancers[10]. The pathway, which p53 controls, normally helps the cell to respond to DNA damage, and then leads to either growth arrest or apoptosis[11–14]. Although in response to genotoxic stress, multiple serine residues on p53 are phosphorylated, p53 activation is not always accompanied by the specific phosphorylation events proposed to be important for its function[15–17].
PI3-K, an upstream effector of serine/threonine kinase Akt (also known as protein kinase B [PKB]), is the major mediator of survival signals that protect cells from apoptosis[18,19]. Large studies have shown that PI3-K provides an anti-apoptotic signal in many types of cells. PKB/Akt, a downstream target of PI3-K, is a key serine/threonine kinase required for the inhibition of apoptosis[20,21]. Moreover, the activation of the PI3-K pathway was capable of protecting from p53-mediated apoptosis[22]. Therefore, PI3-K acting in cellular signaling transduction pathways plays a central role in cancer cell survival.
SIRT1, a nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase, is known to inhibit stress-induced apoptosis. It not only regulates p53 function through deacetylation p53, but also inhibits apoptosis by deacetylating the DNA repair factor Ku70, which is bound with Bax in the cytosol, causing it to sequester Bax away from mitochondria [23]. Either the activation of SIRT1 or inactivation of p53 protects against tumor cell apoptotic stimuli induced by anticancer drugs[24].
The aim of this study was to investigate the effector mechanism of NRP-induced U937 cell death. It was found that NRP-induced U937 cell death is through distinct mechanisms and pathways, including apoptosis and necrosis. Moreover, the inhibition of the PI3-K/Akt signaling pathway enhances NRP-induced apoptosis and contributes to the upregulation of p53 protein expression, indicating that NRP-induced U937 cell death is closely associated with the inactivation of PI3-K/Akt and the activation of p53.
Materials and methods
Materials The NRP (the chemical quality is As2S3) were obtained by the high-energy ball-milling machine and techniques. The PI3-K inhibitor wortmannin and Akt inhibitor KP372-1 were purchased from Sigma (St Louis, MO, USA). The SIRT1 inhibitor sirtinol and Ras inhibitor manumycin A were purchased from Calbiochem (La Jolla, CA, USA). Polyclonal antibodies against caspase-3, Akt, phosphorylated Akt, SIRT-1, p53, phosphorylated p53, and horseradish peroxidase (HRP)-conjuated secondary antibodies (goat antirabbit and goat antimouse) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture The U937 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in RPMI-1640 medium (GIBCO, NY, USA) supplemented with 10% fetal bovine serum (Shengma Yuanheng, Beijing, China), 100 mg/L streptomycin, 100 IU/mL penicillin, and 0.03% L-glutamine, and maintained at 37 ºC with 5% CO2 in a humidified atmosphere.
Cytotoxity assay The U937 cells were incubated at 2×104 cells/well in 96-well plates (NUNC, Roskide, Denmark). The cells were incubated with different concentrations (20~100 µg/mL) of NRP for different time periods, or wortmannin and KP372-1 (Sigma, USA) at given concentrations for 1 h before the introduction of 75 µg/mL NRP for 24 h. Then cell growth was measured with 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay. The percentage of cell growth inhibition was calculated as follows:
Cell death (%)=(A570[control]–A570[NRP])×100/A570(control).
Lactate dehydrogenase activity-based cytotoxicity assays Lactate dehydrogenase (LDH) activity was assessed using a standardized kinetic determination kit (Zhongsheng, Beijing, China). LDH activity was measured in both apoptotic and necrotic cells. The viable cells were collected from the culture medium by centrifugation at 1000×g at 4 ºC for 5 min, and 3000×g for apoptosis. The LDH content from the pellets by centrifugation at 3000×g was used as an index of apoptotic cell death (LDHp)[19]. The LDH released in the culture surpernatant (extracellular LDH [LDHe]) was used as an index of necrotic death. The LDH present in the pellets by centrifugation at 1000×g cells was used as an index of intracellular LDH (LDHi). The percentage of apoptotic and necrotic cell death was calculated as follows:
Apoptosis (%)=LDHp/(LDHp+LDHi+LDHe)×100
Necrosis (%)=LDHe/(LDHp+LDHi+LDHe)×100.
Fluorescence morphological examination Apoptotic morphology was studied by staining the cells with the fluorescent DNA-binding dye acridine orange (AO). The cells were harvested and washed 3 times with phosphate-buffered saline (PBS) after being incubated with 75 or 120 µg/mL NRP, and were then stained with 20 µg/mL AO (Sigma, USA) for 15 min. Then the color and structure of the different cell types were observed under a fluorescence microscope (Olympus, Tokyo, Japan)[25].
Flow cytometric analysis The U937 cells (1×106) were harvested and washed once in cold PBS. The cell pellets were fixed in 70% ethanol and washed in cold PBS. Then the pellets were suspended in 1 mL of propidium iodide (PI) solution containing 50 µg/mL PI, 1 g/L RNase A, and 0.1% (w/v) Triton X-100 in 3.8 mmol/L sodium citrate, followed by incubation on ice under dark conditions for 30 min. The samples were analyzed by a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Western blot analysis The U937 cells were treated with NRP for different time periods. Both adherent and floating cells were collected, and then the Western blot analysis was performed as previously described with some modifications. Briefly, the cell pellets were resuspended in lysis buffer consisting of 50 mmol/L HEPES (pH 7.4), 1% Triton-X 100, 2 mmol/L sodium orthovanadate, 100 mmol/L sodium fluoride, 1 mmol/L edetic acid, 1 mmol/L phenylmethylsulphonyl fluoride (PMSF), 10 mg/L aprotinin (Sigma, USA), and 10 mg/L leupeptin (Sigma, USA) and lysed at 4 ºC for 60 min. After centrifugation at 13000×g centrifugation for 15 min, the protein content of the supernatant was determined by a protein assay reagent (Bio-Rad Laboratories, Hercules, CA, USA). The protein lysates were separated by electrophoresis in 12% SDS-PAGE and blotted onto a nitrocellulose membrane. The proteins were detected using a polyclonal antibody and visualized using antirabbit immunoglobulin G conjugated with peroxidase (HRP) and 3,3’-diaminobenzidine tetrachloride (DAB) as the HRP substrate.
Statistical analysis The data are expressed as mean±SD. Statistical comparisons were made by Student’s t-test. P<0.05 was considered significant.
Results
Preparation of NRP The NRP were prepared with ZJM-20/25 Ball Mill (210th Hospital of the People’s Liberation Army, Dalian, China) in PBS. The particles of the NRP were homogeneously decentralized (Figure 1A, 1B). Diameters above 85% NRP were smaller than 200 nm. The suspension was filtrated with a 0.22 μm membrane and diluted with RPMI-1640.

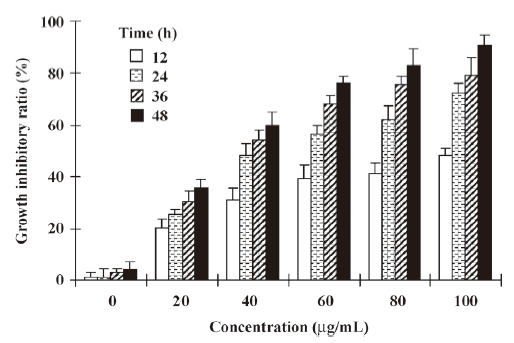
Effect of NRP on the growth of U937 cells The NRP induced U937 cell death in a time- and dose-dependent manner. The incubation of U937 cells with different doses of NRP (20, 40, 60, 80, or 100 µg/mL) for different time periods (12, 24, 36, or 48 h) resulted in a significant increase in the death rate (Figure 2). 24 h after treatment with 75 µg/mL NRP, the cell death rate reached nearly 50% and the IC50 (half number of the cells are dead) at 24 h was 71.7 µg/mL. Therefore, 24 h of incubation with 75 µg/mL NRP is sufficient for the half induction of cell death.
NRP-induced apoptotic cell death in U937 cells To determine the characteristics of NRP-induced U937 cell death, morphological changes were observed using AO staining by fluorescence microscopy. The control cells exhibited uniformly green fluorescence (Figure 3A), while exposure of the U937 cells to 75 µg/mL NRP for 24 h resulted in morphological alterations characteristic of apoptosis, including membrane blebbing, nuclear condensation, and granular apoptotic bodies (Figure 3A).
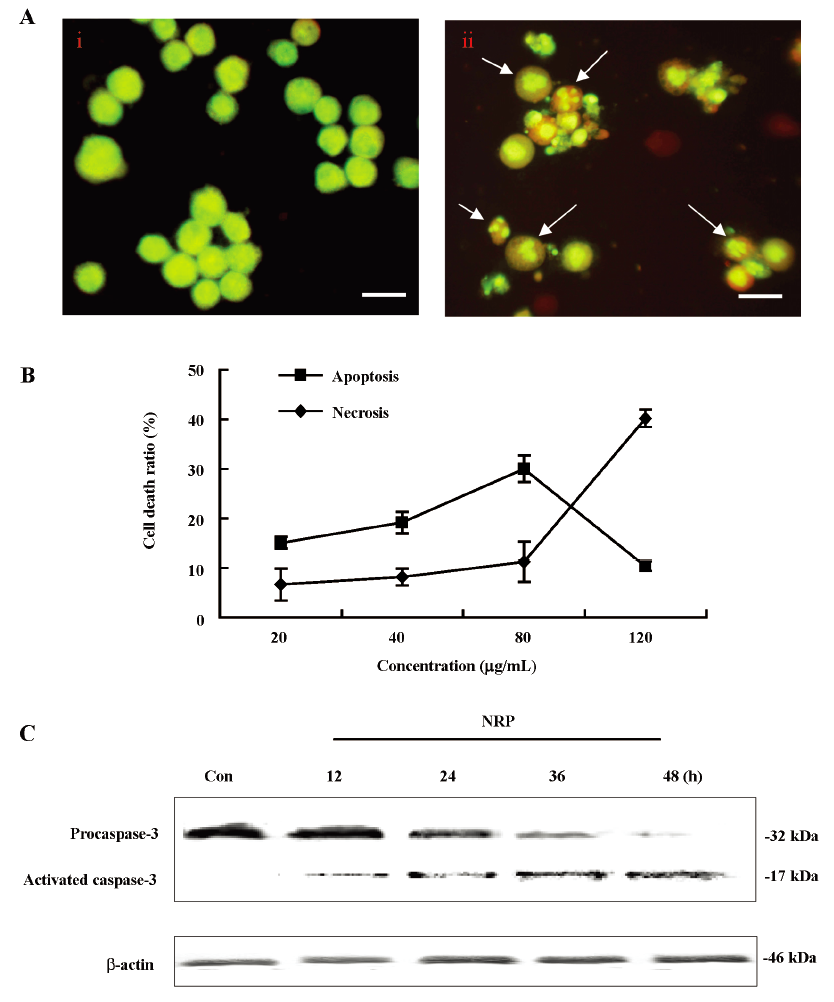
It was reported that the ratio of LDH release from viable cells, apoptosis, and the culture medium might be used to distinguish the number of apoptotic and necrotic cells[26]. In this study, in the presence of NRP (80 µg/mL), the number of apoptotic cells was 30.1% at 24 h; the necrotic cells were still below 10.8%. As the concentration of NRP increased (120 µg/mL), the apoptotic ratio began to decrease with an enhanced necrotic ratio (Figure 3B). Therefore, NRP induced U937 cell death by affecting the balance between apoptosis and necrosis.
However, the activation of caspase-3, another hallmark of apoptosis, was further demonstrated. As the results showed, procaspase-3 was gradually decreased, whereas activated caspase-3 was increased, indicating that NRP time-dependently induced U937 apoptosis (Figure 3C).
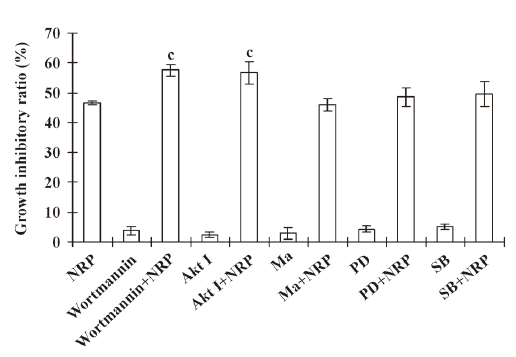
Inactivation of PI3-K contributes to NRP-induced U937 cell death Previous studies showed that the activation of the PI3-K pathway plays an important role in tumor cell proliferation and invasion[18]. Therefore, the U937 cells were pretreated with 200 nmol/L PI3-K inhibitor wortmannin for 1 h, followed by 75 µg/mL NRP treatment. The inhibitory ratio at 24 h was significantly increased compared with the NRP alone-treated group (Figure 4), while Ras inhibitor manumycin A, p38 inhibitor SB203580, and ERK inhibitor PD98059 failed to enhance NRP-induced growth-inhibitory effects in U937 cells. These results indicated that the inhibition of PI3-K contributed to NRP-induced cell death.
To further confirm the above results, a flow cytometric analysis was performed (Figure 5). It was found that in the presence of 75 µg/mL NRP, the apoptotic ratio (the percentage of cells in sub-G0/G1 phase, a hallmark of apoptosis) reached 26.6% (Figure 5D) from the baseline of the control (2.62%; Figure 5A), whereas pretreatment with wortmannin significantly increased the apoptotic ratio (35.9%; Figure 5E) compared with the group treated by NRP alone.
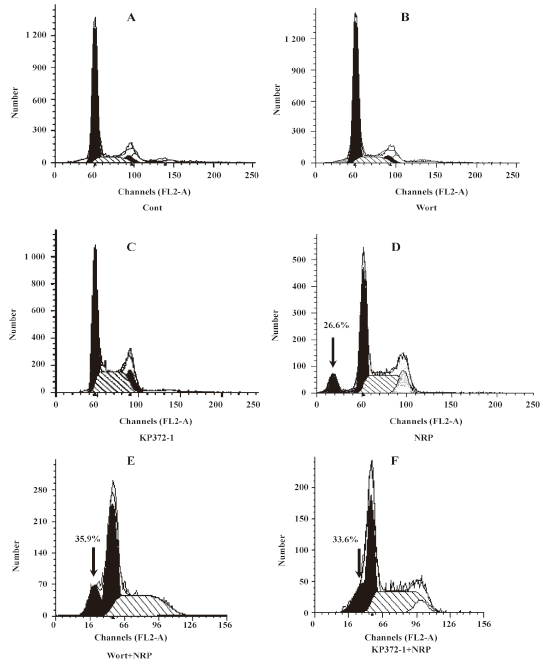
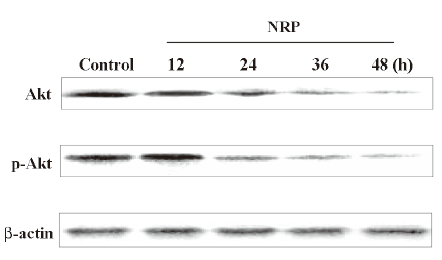
Effect of Akt on NRP-induced U937 apoptosis Activated PI3-K converts phosphatidylinositol 4,5 phosphate into phosphatidylinositol 3,4,5 phosphate, which results in the recruitment of Akt and the phophorylation of PI3-K at residues T308 and S473[19]. Here, the U937 cells were cultured with NRP for 24 h after being pretreated with 10 µmol/L Akt inhibitor KP372-1 for 60 min, and the ratio of cell death was detected (Figure 4). Consistent with the result of the pre-incubation of wortmannin, KP372-1 promoted NRP-induced U937 cell death, and the apoptotic ratio increased from 26.6% to 33.6% (Figure 5F).
Then the Western blot analysis was performed to detect the protein expressions of Akt and p-Akt. As shown in Figure 6, Akt and Akt phosphorylation were reduced by NRP treatment and this suppressive effect became more significant with the extension of the incubation time with NRP.
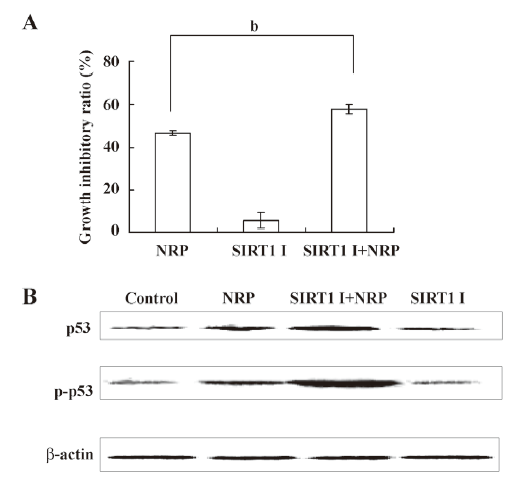
Inactivation of SIRT1 contributes to p53 activation in NRP-induced U937 cell apoptosis SIRT1 has been shown to be associated with apoptosis, and the expression of the inactive SIRT1 protein can potentiate p53-mediated apoptosis[27,28]. In order to demonstrate whether such a mechanism is involved in NRP-induced cell apoptosis, SIRT1 inhibitor sirtinol was pretreated for 1 h before NRP was introduced. After 24 h, the growth inhibitory ratio was detected by MTT. It was found that the inactivation of SIRT1 facilitates NRP-induced U937 cell death (Figure 7A). In order to further confirm this mechanism, a Western blot analysis was performed to detect the expressions of p53 and p-p53. As shown in Figure 7B, the expression of p53 and its phosphorylation were upregulated by NRP treatment, and the inhibition of SIRT1 activity with sirtinol contributed to the activation of p53.
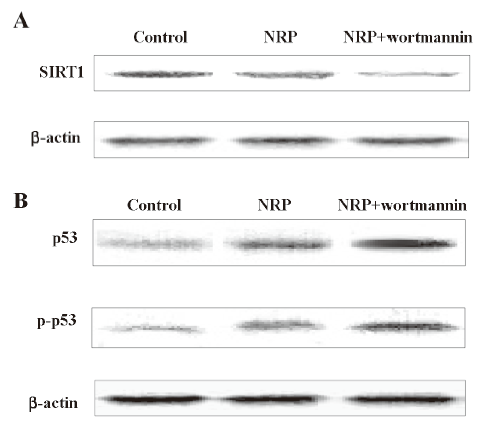
Decreased expression of SIRT1 and p53 activation were dependent on PI3-K/Akt inactivation The cells were pretreated with 200 nmol/L wortmannin for 1 h, followed by the addition of 75 µg/mL NRP. After 24 h, the cell lysates were separated by 10% SDS-PAGE, and SIRT1, p53, and the phosphorylated p53 protein bands were detected by Western blot analysis. The results showed that wortmannin pretreatment executed a more significant inhibitory effect on the expression of SIRT1, as well as more marked augmented effect on p53 activation, compared with the NRP-alone treatment group (Figure 8).
Discussion
In this study, we demonstrated that NRP inhibits U937 cell growth and induces apoptosis. These biochemical events might be associated with the inhibition of the PI3-K/Akt signaling pathway, the inactivation of SIRT1, and the activation of the p53 tumor suppressor gene. Importantly, the inhibition of PI3-K activity significantly augmented NRP-induced U937 cell apoptosis, leading to the downregulated expression of the SIRT1 protein and the activation of p53.
It is well known that the majority of death signals pass through 2 distinct pathways: apoptosis and necrosis[29]. Some signaling pathways, such as death receptors, mitogen-activate protein kinase, and caspase cascades, participate in both processes, and it is possible to switch apoptosis to necrosis by regulating the signaling pathways[30]. In this study, we demonstrated that NRP inhibited the proliferation of U937 cell in vitro in a dose- and time-dependent manner. There were significant growth inhibitory effects at 20–100 µg/mL NRP. Based on the changes of cellular morphology, the LDH activity-based assay, and the activation of caspase-3, we concluded that NRP induced U937 cell death by mediating the balance between apoptosis and necrosis.
The enzyme PI3-K is composed of a 110 kDa catalytic subunit and an 85 kDa adaptor subunit. When growth factors, such as insulin, insulin-like growth factor-1, fibroblast growth factor-2, and others bind to their cognate cell-membrane receptor, the p85 subunit is recruited to the C-terminal (intracellular) part of the growth factor receptor. Subsequent dimerization with the P110 unit then leads to the full enzymatic activity of PI3K[31]. Accumulating evidence suggests that PI3-K is an important regulator of cell survival. Downstream of PI3-K lies PKB/Akt and PLCγ, which stimulate the process of cell proliferation[32]. Our results show that PI3-K inhibitor wortmannin, and Akt inhibitor KP372-1, significantly augmented NRP-induced apoptosis, while other signaling molecule inhibitors, such as the Ras inhibitor manumycin A, ERK inhibitor PD98059, and P38 inhibitor SB203580 did not show these apoptosis-augmented effects. NRP downregulated the expressions of Akt and p-Akt, indicating that the PI3-K/Akt signaling cascade might be partially involved in the apoptosis-inducing effects of NRP in U937 cells.
The p53 protein is a regulator of cell cycle progression and a mediator of apoptosis in many cell lines. A large body of data documented that the p53 gene is often mutated in many tumor cells and the mutations contribute to clonal cellular expansion and genomic instability because of decreased DNA repair and apoptosis[33,34]. MDM2 (transformed 3T3 cell double minute 2) is an E3 ligase, which can ubiquitylate the lysine residues at the C terminus of p53[35–37]. It inhibits p53 using at least 2 mechanisms, namely, by ubiquitination of p53 on the lysine residues at its C terminus, leading to proteosomal degradation, and by blocking a transactivation domain of p53[38,39]. Here in our experiments, both the total p53 and p-p53 protein levels increased. It is possible that there may be a portion of total p53 becoming acetylated under the condition of NRP treatment, leading to the upregulation of p-p53 protein expression. At the same time, a part of ubiquitylated p53 was downregulated. Therefore, the result of the balance between acetylation and ubiquitylation in this system showed that total p53 increased.
Moreover, MDM2 also serves as a good substrate for Akt. A recent study showed that the PI3-K/Akt signaling pathway inhibited p53-mediated transcription and apoptosis through inducing the phosphorylation of MDM2 and enhancing the degradation of p53[40]. In the present study, Akt and p-Akt were downregulated, p53 and p-p53 were correspondingly upregulated. The inhibition of PI3-K activity contributed to the upregulated expressions of the p53 and p-p53 proteins and increased the ratio of cell death. These phenomena suggest that a tight association between the PI3-K/Akt signaling pathway and p53 in NRP-induced U937 cell apoptosis exists. Covering this pathway will be essential in understanding how p53 acetylation is regulated, and how Akt regulates its function is a very important subject of our study, so our further study will concentrate on this aspect.
SIRT1 is a key regulator involved in cell aging and response to DNA damage and is a member of the sirtuin family of NAD+-dependent deacetylase known to inhibit stress-induced apoptosis[26,41]. It also regulates p53 function via deacetylation with a specificity for the p53 C-terminal Lys382 residue, a modification of which has been implicated in the activation of p53 as a transcription factor[27]. Our present results show that 75 µg/mL NRP abrogates SIRT1 expression while facilitating p53 activation. Moreover, the inhibition of PI3-K activity further attenuates SIRT1 expression by the administration of wortmannin. These results indicate that the inactivation of SIRT1 might be in part associated with PI3-K inactivation-induced cell death in NRP-treated U937 cells.
Noticeably, the inhibition of SIRT1 activity by using SIRT1 inhibitor sirtinol augmented p53 activation. These results indicate that the activation of p53 was due to the downregulated deacetyl-function of SIRT1 induced by the inhibition of the PI3-K/Akt signaling pathway in NRP-treated U937 cells. However, further study will be required to elucidate the relationship of SIRT1 and p53 in the apoptotic action of NRP in U937 cells.
NRP induced U937 cell apoptosis and inhibited cell growth in a time- and dose-dependent manner. The mechanism is in part due to the inhibition of the PI3-K/Akt signaling pathway. The potential effects of wortmannin on NRP-induced cell death reflect that the inhibition of the PI3-K survival pathway is necessary for the SIRT1 inactivation and p53 activation in NRP-induced U937 cell apoptosis.
References
- Luo LY, Huang J, Gou BD, Zhang TL, Wang K. Induction of human promyelocytic leukemia HL-60 cell differentiation into monocytes by arsenic sulphide: Involvement of serine/threonine protein phosphatases. Leuk Res 2006;30:1399-405.
- Wu JZ, Ho PC. Evaluation of the in vitro activity and in vivo bioavailability of realgar nanoparticles prepared by cryo-grinding. Eur J Pharm Sci 2006;29:35-44.
- Ning N, Peng ZF, Yuan L, Gou BD, Zhang TL, Wang K. Realgar nano-particles induce apoptosis and necrosis in leukemia cell lines K562 and HL-60. China J Chin Mat Med 2005;30:136-40. Chinese..
- Kim SO, Han J. Pan-caspase inhibitor zVAD enhances cell death in RAW 246.7 macrophages. J Endotoxin Res 2001;7:292-6.
- Kawazoe N, Watabe M, Masuda Y, Nakajo S, Nakaya K. Tiamil is involved in the regulation of bufalin-induced apoptosis in human leukemia cells. Oncogene 1999;18:2413-21.
- Hill PA, Tumber A, Meikle MC. Multiple extracellular signals promote osteoblast survival and apoptosis. Endocrinology 1997;138:3849-58.
- Mizukami S, Kikuchi K, Higuchi T, Urano Y, Mashima T, Tsuruo T, et al. Imaging of caspase-3 activation in HeLa cells stimulation with etoposide using a novel fluorescent probe. FEBS Lett 1999;453:356-60.
- Wu Z, Wu LJ, Li LH, Tashiro SI, Onodera S, Ikejima T. p53-mediated cell cycle arrest and apoptosis induced by shikonin via a caspase-9-dependent mechanism in human malignant melanoma A375-S2 cell death. J Pharmacol Sci 2004;94:166-76.
- Qiao AM, Ikejima T, Tashiro S, Onodera S, Zhang WG, Wu YL. Involvement of mitochondria and caspase pathways in N-demethyl-clarithromycin-induced apoptosis in human cervical cancer HeLa cell. Acta Pharmacol Sin 2006;27:1622-9.
- Crow MT. Revisiting p53 and its effectors in ischemic heart injury. Cardiovasc Res 2006;70:401-3.
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ 2003;10:431-42.
- Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer 2002;2:594-604.
- Ciciarello M, Mangiacasale R, Casenghi M, Limongi MZ, D’Angelo M, Soddu S, et al. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J Biol Chem 2001;276:19205-13.
- Karpinich NO, Tafani M, Rothman RJ, Russo MA, Farber JL. The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochrome c. J Biol Chem 2002;277:16547-52.
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997;91:325-34.
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998;281:1677-9.
- Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 2000;287:1824-7.
- Lee TK, Man K, Ho JW, Sun CK, Ng KT, Wang XH, et al. FTY720 induces apoptosis of human hepatoma cell lines through PI3-K-mediated Akt dephosphorylation. Carcinogenesis 2004;25:2397-405.
- McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Advan Enzyme Regul 2006;46:249-79.
- Kalkman HO. The role of the phosphatidylinositide 3-kinase–protein kinase B pathway in schizophrenia. Pharmacol Therapeut 2006;110:117-34.
- Trtoman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumor suppressor network opposing nuclear Akt function. Nature 2006;441:523-7.
- Zhang CL, Wu LJ, Zuo HJ, Tashiro SI, Onodera S, Ikejima T. Cytochrome c release from oridonin-treated apoptosis A375-S2 cells is dependent on p53 and extracellular signal-regulated kinase activation. J Pharmacol Sci 2004;96:155-63.
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004;305:390-2.
- Minamino T, Komuro I. Hyperglycemia-induced endothelial senescence mediated by the p53 deacetylase SIRT1 contributes to diabetic vasculopathy. Vascul Pharmacol 2006;45:e93.
- Mao YE, Liu JK, Lu ZX, Yan Z, Liu SF, Li LL, et al. Grifolin, a potential antitumor natural product from the mushroom Albatrellus confluens, inhibits tumor cell growth by inducing apoptosis in vitro. FEBS Lett 2005;579:3437-43.
- Zhang CL, Wu LJ, Tashiro SI, Onodera S, Ikejima T. Oridonin induces apoptosis of HeLa cells via altering expression of Bcl-2/Bax and activating caspase-3/ICAD pathway. Acta Pharmacol Sin 2004;25:691-8.
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004;430:686-9.
- Vaziri H, Dessain SK, Ng EE, Imai SI, Frye RA, Pandita TK, et al. Hsir2 (SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001;107:149-59.
- Vercammen D, Beyaet R, Denecker G, Goossens V, Van Loo G, Declercq W, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 1998;187:1477-85.
- Zhang CL, Wu LJ, Tashiro SI, Onodera S, Ikejima T. Oridonin induces a caspase-independent but mitochondria-and MAPK dependent cell death in the murine fibrosarcoma cell line L929. Biol Pharm Bull 2004;27:1527-31.
- Kalkman HO. The role of the phosphatidylinositide 3-kinase–protein kinase B pathway in schizophrenia. Pharmacol Therapeut 2006;110:117-34.
- Liu YQ, You S, Tashiro SI, Onodera S, Ikejima T. Activation of phosphoinositide 3-kinase, protein kinase C, and extracellular signal-regulated kinase is required for oridonin-enhanced phagocytosis of apoptotic bodies in human macrophage-like U937 cells. J Pharmacol Sci 2005;98:361-71.
- Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu LJ, et al. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci USA 1999;96:3706-11.
- Ford J, Jiang M, Milner J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res 2005;65:10457-63.
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is an ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 1997;420:25-7.
- Nakamura S, Roth JA, Mukhopadhyay T. Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2- dependent protein degradation and ubiquitination. Mol Cell Biol 2000;20:9391-8.
- Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT. Multiple C-terminal lysine residues target p53 for ubiquitinproteasome-mediated degradation. Mol Cell Biol 2000;20:8458-67.
- Mayo LD, Dixon JE, Durden DL, Tonks NK, Donner DB. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J Biol Chem 2002;277:5484-9.
- Mayo LD, Donner DB. The PTEN, Mdm2, p53 tumor suppressoroncoprotein network. Trends Biochem Sci 2002;27:462-7.
- Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, et al. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem 2002;277:21843-50.
- Cui Q, Tashiro SI, Onodera S, Ikejima T. Augmentation of oridonin-induced apoptosis observed with reduced autophagy. J Pharmacol Sci 2006;101:230-9.