
Agonist-induced hump current production in heterologously-expressed human α4β2–nicotinic acetylcholine receptors1
Introduction
Nicotinic acetylcholine receptors (nAChR) are prototypes of the ligand-gated ion channel superfamily of neurotransmitter receptors[1–4]. nAChR have historic importance, as their existence as “receptive substances” in vertebrate muscles was gleaned a century ago[5]. They have become models for the establishment of concepts pertaining to mechanisms of drug action, synaptic transmission, and the structure and function of transmembrane signaling molecules. Several nAChR subtypes have distinctive features that are dictated in part by their composition from subunits derived from at least 16 genes. The predominant, high-affinity, nicotine-binding nAChR subtype in the brain contains α4 and β2 subunits (α4β2–nAChR)[1,6]. Numbers and/or functions of α4β2–nAChR are affected by nicotine at concentrations found in the plasma of tobacco users[1,7–9], and α4β2–nAChR have been implicated in nicotine self-administration, reward, and dependence[1,2,10,11]. α4β2–nAChR also play important roles in health and a variety of neuropsychiatric diseases[12].
Like acetylcholine (ACh), nicotine acts acutely to cause rapid opening of nAChR channels. However, these responses are transient, and channel opening frequency diminishes with protracted exposure to high concentrations of agonists through a process (or a series of processes) termed “desensitization”[13,14]. Depending on the duration and concentration of agonist exposure, the rates of recovery from desensitization can vary, but explanations of mechanisms involved in the induction and recovery from desensitization have been elusive. Studies done mostly using muscle-type nAChR suggest that open-channel block by a high concentration of agonists contributes to the loss of function, and evidence for this has come from work showing the production of a transient nAChR response after the removal of applied agonists called a “hump” or “rebound” current[15,16]. Hump currents also have been observed for other channels, and one interpretation has been that they reflect the transient reactivation of channel opening when agonist molecules that had engaged in open-channel block leave the channel pore[16–18].
Here we report on experiments that tested the hypothesis that open-channel block by high agonist concentrations and hump current production are attributes of human α4β2–nAChR.
Materials and methods
Subclonal human epithelial 1 (SH-EP1)–hα4β2 cells Established techniques[19] were used to introduce human α4 (S452) and β2 subunits (kindly provided by Dr Ortrud STEINLEIN, Institute of Human Genetics, University Hospital, Ludwig-Maximillians-University, Germany) and subcloned into pcDNA3.1-zeocin and pcDNA3.1-hygromycin vectors, respectively) into native nAChR-null SH-EP1 cells[20] to create the stably-transfected, monoclonal SH-EP1-hα4β2 cell line heterologously expressing human α4β2–nAChR. Cell cultures were maintained at low passage numbers (1–26 from our frozen stocks to ensure the stable expression of the phenotype) in complete medium[21] augmented with 0.5 mg/mL zeocin and 0.4 mg/mL hygromycin (to provide a positive selection of transfectants) and passaged once weekly by splitting the just-confluent cultures 1/20 to maintain cells in proliferative growth. RT–PCR, immunofluorescence, radioligand-binding assays, and isotopic ion flux assays were conducted recurrently to confirm the stable expression of α4β2–nAChR as message-, protein-, and ligand-binding sites, and functional receptors.
Patch-clamp whole-cell current recordings Conventional whole-cell current recording, coupled with computer-controlled U-tube fast application and the removal of agonists, was applied in this study as previously described[22–24]. Briefly, the cells plated on polylysine-coated 35 mm culture dishes were placed on the stage of an inverted microscope (Olympus iX70, Lake Success, NY, USA) and continuously superfused with standard external solution (2 mL/min). Glass microelectrodes (3–5 MΩ resistance between the pipette and extracellular solutions) were used to form tight seals (>1 GΩ) on the cell surface until suction was applied to convert to conventional whole-cell recording. The cells were then voltage-clamped at holding potentials of –60 mV, and ion currents in response to application of ligands were measured (Axon Instruments 200B amplifier, Foster City, CA, USA). Whole-cell access resistance was less than 20 MΩ before series resistance compensation. Both pipette and whole-cell current capacitances were minimized, and series resistance was routinely compensated to 80%. Typically, data were acquired at 10 kHz, filtered at 2 kHz, displayed and digitized online (Digidata 1322 series A/D board, Axon Instruments, USA), and stored to a hard drive. Data acquisition of whole-cell currents was done using Clampex 9.2 (Axon Instruments, USA), and the results were plotted using Origin 5.0 (Microcal, North Hampton, MA, USA) or Prism 3.0 (GraphPad Software, San Diego, CA, USA). nAChR acute desensitization (the decline in inward current amplitude over the course of agonist application) was analyzed for decay half-time (τ=0.693/k for decay rate constant k), peak current (Ip), and steady-state current (Is), using fits to the mono (or double) exponential expression I=([Ip–Is] e–kt)+Is (or I=([Ip–Ii] e–k1t)+([Ii–Is] e–k2t)+Is, where Ii is the intermediate level of current and k1 and k2 are rate constants for the 2 separate decay processes. Curve fitting usually was done using data between 90% and 10% of the difference between peak and steady-state currents. The experimental data are presented as mean±SEM, and comparisons of different conditions were analyzed for statistical significance using t-tests. All experiments were performed at room temperature (22±1 oC). Concentration response profiles were fit to the Hill equation and analyzed using Origin 5.0.
Patch-clamp single-channel recordings Outside-out patch, single-channel recordings were performed to compare single-channel properties of α4β2–nAChR-mediated currents at different concentrations of ACh or at different holding potentials. After establishing conventional whole-cell configuration, the recording electrode was gently removed from the cell and an outside-out patch was formed. Single-channel signals activated by application of nicotine via a U-tube were acquired while being filtered at 1 kHz (8-pole Butterworth filter and digitized at 10 kHz; Digidata interface 1322A). The results were analyzed using Clampfit 9.2. Time constants for open and closed intervals and amplitude distributions were computed and corrected for dead time (defined as the longest duration event that was allowed to be missed; usually 250 ms). Channel amplitude histograms were constructed from open-time events with durations longer than the dynamic frequency response limit of the entire system in order to exclude amplitude attenuation due to system bandwidth limitation. Channel open-time histograms were based on open events with a duration longer than 0.2 ms separated by closed times no shorter than 0.2 ms. Open-time constants (t) were obtained from least squares fits of the data; the mean open time (to) was calculated as the arithmetic mean of the sum of all open events within a record. In addition, records were analyzed for open events shorter than or equal to 1 ms as a percentage of all open events. Channel conductance distribution, the mean open time, and the mean closed time were fit using Qub software (State University of New York, Buffalo, NY, USA).
Homology modeling and ACh docking into channel pore Five sets of 4 transmembrane domains of the human nicotinic receptor subunits with a subunit arrangement of α4β2β2α4β2 (clockwise from an extracellular view) with chain breaks were aligned with the deduced structure of transmembrane domains from the Torpedo nicotinic receptor (protein data bank file 1OED) using the modeler in Discovery Studio 1.7 (Accelrys, San Diego, CA, USA; “Align Sequence with Structure” protocol with blosum62 scoring matrix, gap open penalty of –200, gap extension penalty of –10, and default 2-D gap weights). The homology model was then built using the “Building Homology Models” protocol. The resulting model was further typed with the “CHAMm force field” tool and energy minimized by the “minimization” protocol with 400 steps of steepest descent cycles followed by 1000 steps of conjugate gradient algorithm. The docking of ACh into the ion channel pore of the α4 and β2 subunit homology model was performed with ICM pro (Molsoft, San Diego, CA, USA) with manual adjustment of the docking box to cover the full length of the pore region. The docking result was presented with Swiss PDB Viewer 3.7 (Swiss Institute of Bioinformatics, Basel, Switzerland) and rendered by POV-Ray 3.6 (Persistence of Vision Raytracer Pty Ltd, Williamstown, Victoria, Australia).
Solutions and drug application The standard external solution contained (in mmol/L): 120 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 25 D-glucose, and 10 HEPES, and was adjusted to pH 7.4 with Tris-base. In the experiments, ACh was applied as an agonist without atropine since our preliminary data showed that 1 µmol/L atropine sulfate did not affect ACh-induced currents[22], and also atropine itself was reported to block nAChR[25]. For most conventional whole-cell recordings, K+ electrodes were used and filled with solution containing (in mmol/L): 140 KCl, 4 MgSO4, 0.1 EGTA, 4 Mg-ATP, and 10 HEPES, and adjusted to pH 7.2 with Tris-base. In other experiments, Tris+ electrodes were used and filled with solution containing (in mmol/L): 110 Tris phosphate dibasic, 28 Tris-base, 11 EGTA, 2 MgCl2, 0.1 CaCl2, and 4 Mg-ATP; pH 7.3[26]. For studies in Ca2+-free external solutions, external 2 mmol/L CaCl2 was replaced with 4 mmol/L NaCl, and 1 mmol/L EGTA was added to the solution.
To initiate whole-cell current responses, nicotinic agonists were delivered into the bath medium near to the cell being recorded via a computer-controlled U-tube system so that solution exchange occurred within 30 ms (based on 10%–90% peak current rise times). Intervals between drug applications (3 min) were adjusted specifically to ensure the stability of nAChR responsiveness (without functional rundown), and the selection of pipette solutions used in most of the studies described here was made with the same objective. The drugs used in the present study were (–) nicotine, ACh, dimethyl-phenyl-piperazinium (DMPP), epibatidine (EPBD), cytisine, lobeline, and dihydro-β-erythroidine (DHβE); all were purchased from Sigma (St Louis, MO, USA).
Results
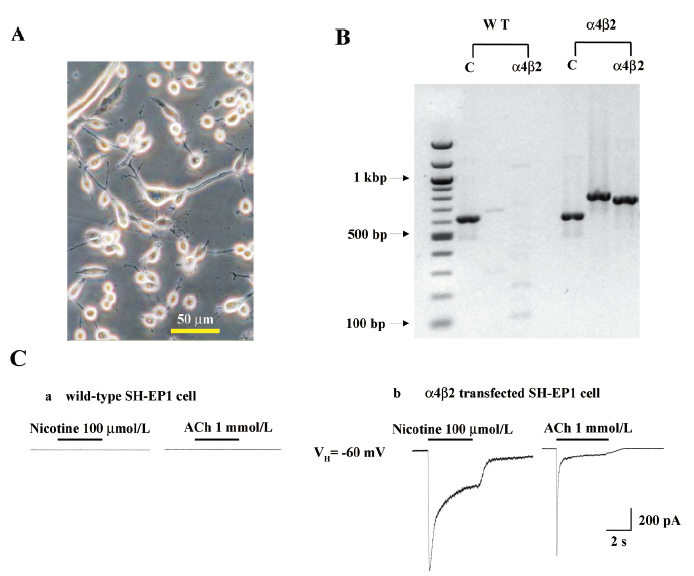
Heterologous expression of human α4β2–nAChR in SH-EP1 cells SH-EP1 cells exhibited a range of morphologies before (data not shown) or after (Figure 1A) transfection with nAChR α4 and β2 subunits. The RT–PCR analyses showed the expression of human nAChR α4 and β2 subunit messages in the SH-EP1–hα4β2 cells (Figure 1B). In contrast, there was no such expression in the absence of the reverse transcription step or in the untransfected cell host despite successful amplification of GAPDH message in all of the cells (Figure 1B). Apparent levels of α4 and β2 subunit transcripts assessed using mRNA fluorescence in situ hybridization did not seem to fluctuate as a function of cell confluence or between grouped or solitary cells (data not shown). Functionally, nicotinic agonists (nicotine and ACh) failed to induce detectable currents in the untransfected cells (Figure 1C), but induced inward currents in α4- and β2-transfected cells (Figure 1C). These results indicated the appropriate expression of nAChR subunit cDNA as messages in stably-transfected SH-EP1 cells and the generation of functional nAChR as a consequence.
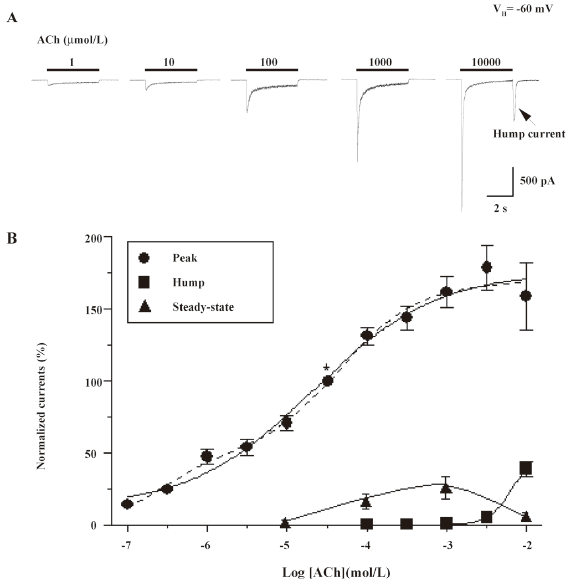
Concentration-dependence of hump current production Open-channel block by nicotinic agonists at high concentrations has been previously reported for several muscle-type nAChR, and one of its manifestations is the production of hump currents[16,27,28]. Our initial studies examined whole-cell current responses of human α4β2–nAChR stably expressed in SH-EP1–hα4β2 cells to ACh applied as 4 s pulses at 3 min intervals at concentrations between 1 µmol/L and 10 mmol/L (Figure 2A). The peak whole-cell current responses of α4β2–nAChR to nicotine or ACh were achieved rapidly after the application of the agonist and showed a concentration dependence that was well fitted to the Hill equation, yielding a half-maximal effective concentration (EC50) and a Hill coefficient of 21 µmol/L and 0.6 for ACh (see figure 2 legend for fits to 1- or 2-site models). In this study, we defined whole-cell current responses that typically decay during ACh exposure to approach steady-state levels as acute desensitization (ie loss of function during acute exposure to agonists) of α4β2–nAChR function. The dose dependence for the absolute magnitude of steady-state currents was bell-shaped because steady-state inward current amplitudes fell from their highest levels at intermediate concentrations (~100 µmol/L) of ACh to lower levels at or above 1 mmol/L ACh (Figure 2B). Inward hump currents evident during drug washout were observed for whole-cell currents induced by either nicotine (1 mmol/L) or ACh (10 mmol/L), and the concentration dependence for this effect had features of the early phase of a sigmoid log concentration–response profile (Figure 2B).
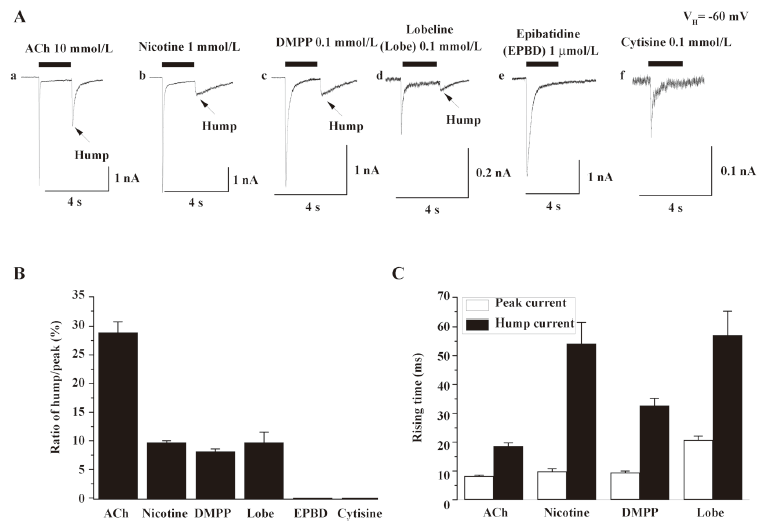
Hump currents induced by different agonists To determine whether nicotinic agonists differ in their abilities to induce hump currents, whole-cell current responses were ascertained for several agonists applied at concentrations near to those that produced maximal responses of human α4β2–nAChR[19,24]. Whole-cell current responses from 6–10 cells showed that hump currents were induced by 10 mmol/L ACh, 1 mmol/L nicotine, 0.1 mmol/L DMPP, and 0.1 mmol/L lobeline, but not by 1 μmol/L EPBD or 0.1 mmol/L cytisine. Figure 3 summarizes the ratio of hump/peak currents induced by different agonists (Figure 3B) and the rising time of peak and hump currents induced by 10 mmol/L ACh, 1 mmol/L nicotine, 0.1 mmol/L DMPP, and 0.1 mmol/L lobeline. This finding discounted the possibility of a technical artifact due to the poor removal of drug and/or wash-back reperfusion of the cell with agonists because not all agonists produced hump currents. ACh, nicotine, DMPP, and EPBD are full agonists, whereas cytisine and lobeline are partial agonists at human α4β2–nAChR, indicating that hump current production was not an attribute of only full or partial agonists. The rates of acute desensitization were faster for ACh, nicotine, EPBD, and lobeline than for DMPP or cytisine, indicating that hump current production was not obviously related to the acute desensitization rate. ACh and DMPP are positively-charged, quaternary ammonium ions, whereas the other compounds are not. Lobeline and ACh are the largest and smallest of these compounds, respectively. Thus, the ability to induce hump currents is not simply and solely attributable to agonist size, charge, or acute functional potency.
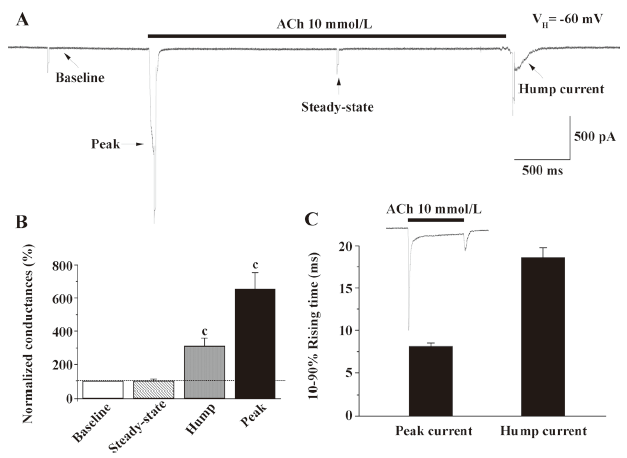
Conductance change associated with hump current production When hyperpolarizing pulses (10 mV, 50 ms) were applied at different times during the recording of whole-cell current responses to monitor cellular membrane conductance, evidence for conductance increases proportional to inward current amplitude was obtained during both peak and hump current phases (Figure 4). In contrast, conductance during the steady-state phase of the whole-cell response was similar to that prior to the application of agonists, suggesting that the receptor desensitized to a non-functional status.
Current-voltage relationship for hump currents To test the hypothesis that hump currents reflect the reactivation of nAChR recovering from open-channel block after agonist removal, current-voltage relationships for peak and hump currents were assessed. When whole-cell current responses to 10 mmol/L ACh were measured using cells voltage clamped at different holding potentials, both peak and hump current responses showed similar current-voltage relationships and reversal potentials (~0 mV) and evidence for inward rectification (Figure 5A–5C). These findings are consistent with current transit through the same channel during peak or hump currents mediated by α4β2–nAChR.
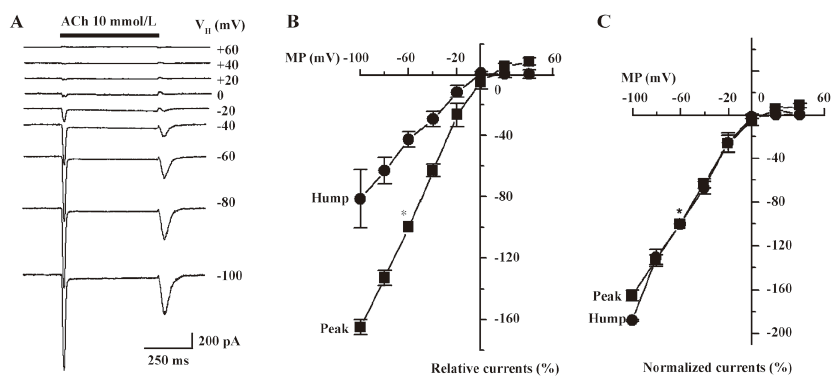
Pharmacological blockade of hump currents To further characterize the pharmacological features of hump currents, the effects of DHβE were determined. DHβE is a potent antagonist at α4β2–nAChR, as it reduces whole-cell peak current responses to 3 µmol/L nicotine with a half-maximal inhibitory concentration (IC50) of –6.05±0.03 mol/L and a Hill coefficient of –1.45±0.16 (Figure 6A). DHβE appears to operate as a competitive antagonist[19] because its presence at a concentration of 300 nmol/L shifted nicotine-induced peak current response curves to the right (the nicotine EC50 of 3.1 µmol/L in the absence of DHβE was shifted to 9.2 µmol/L in the presence of 300 nmol/L DHβE; Figure 6B). The effects were measured on whole-cell current responses to 10 mmol/L ACh when 100 µmol/L DHβE was applied at different times relative to the application of agonists (Figure 6C). Under conditions where ACh application alone induced both peak and hump currents (Figure 6C), DHβE application simultaneous with ACh application dramatically reduced peak current responses and eliminated hump currents (Figure 6C). Brief pretreatment with DHβE, followed by exposure to both ACh and continuing DHβE, fully blocked both peak and hump currents (Figure 6C). If DHβE was applied after peak current production by ACh, but before ACh was washed out, a complete block of the hump currents occurred (Figure 6C). If DHβE application preceded ACh application, the peak current responses were blocked, but if the removal of DHβE occurred prior to ACh washout, hump current production became evident (Figure 6C). These results indicate that DHβE can block both peak and hump currents or can block either peak or hump currents separately, depending on the timing of drug applications, which is consistent with the production of hump currents by reactivation of α4β2–nAChR after the washout of agonists applied at high concentrations.
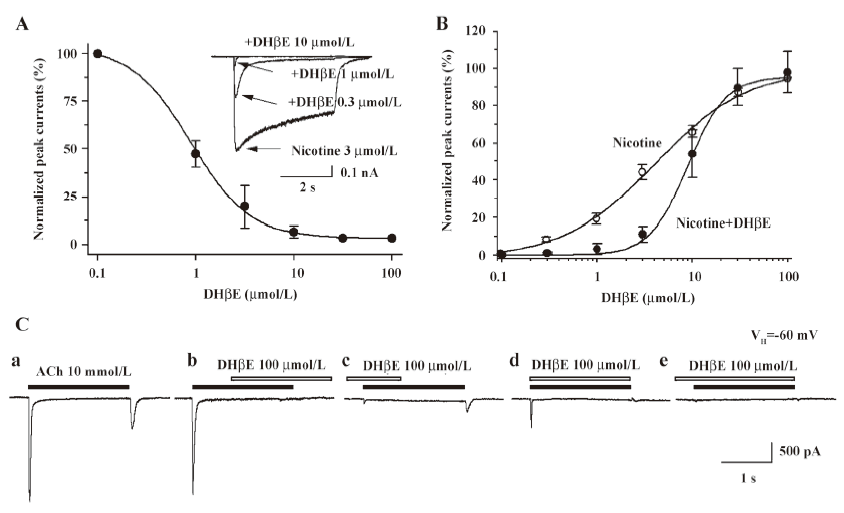
Hump current amplitude is smaller for desensitized nAChR Acute desensitization of nAChR responses during seconds of agonist exposure is reversible if nAChR are allowed to recover before being challenged again with agonists. However, if agonist exposure continues, then recovery from loss of function is slower, apparently reflecting conversion to more deeply desensitized states. To test for the sensitivity of hump current production to nAChR desensitization, 2 approaches were applied to manipulate the rates and extents of desensitization and to determine the consequences for hump current production. First, the ability to manipulate the rates and extents of nAChR desensitization by conducting whole-cell current recording using pipettes filled with different solutions was exploited. The rates and extents of acute desensitization occurring during agonist exposure were higher if whole-cell current recordings were derived using K+ electrodes, rather than using Tris+ electrodes (Figure 7A). When peak whole-cell current amplitudes in response to 10 mmol/L ACh were normalized, the magnitudes of hump currents also were lower when recorded using K+ electrodes than when using Tris+ electrodes (Figure 7A). Thus, the higher rate and greater extent of nAChR desensitization observed when the cytosolic space was perfused with K+ electrode solution was associated with eliminated hump current magnitude. In contrast, a lower rate and extent of desensitization occurred when recordings were made using Tris+ electrodes, and more nAChR were available to participate in hump current production. Second, the ability to manipulate levels of nAChR desensitization by altering the duration of ACh exposure was exploited. When normalized to peak whole-cell current response to 10 mmol/L ACh, hump current amplitude was greatest for the shortest duration of ACh exposure and smallest for the longest ACh exposure time (Figure 7B). Figure 7B shows that hump currents declined with prolonged agonist exposure time. This suggests that desensitization of nAChR also desensitizes hump current production, consistent again with hump current production by nAChR capable of responding to agonists during removal from blocked channels.
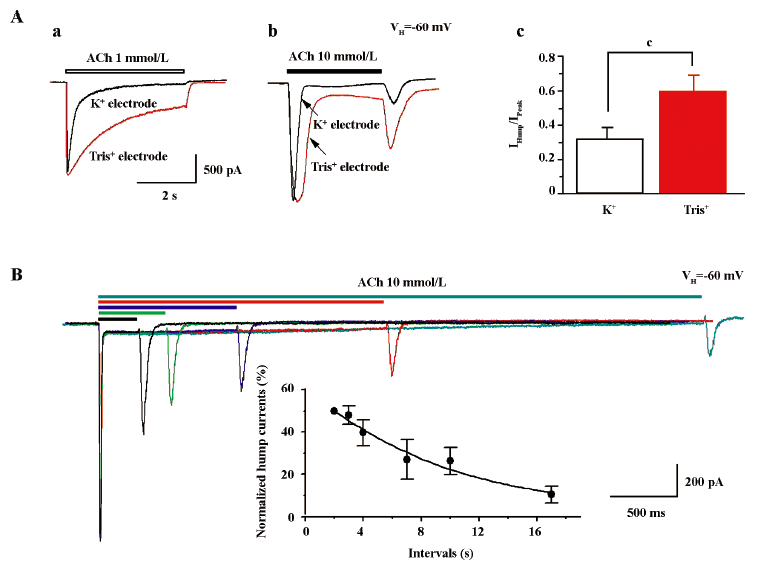
Extracellular Ca2+ eliminates hump currents We altered external Ca2+ concentrations to ascertain the effects on hump current production. At a concentration of 1 mmol/L ACh, little hump currents were induced when the external medium contained 2 mmol/L Ca2+ (Figure 8A), while obvious hump currents were produced after external solution exchange to remove extracellular Ca2+ (ie using medium that contained no Ca2+, but was supplemented with 1 mmol/L EGTA; Figure 8A). Figure 8B summarizes the effects of external Ca2+ on ACh-induced currents. Across 7 tested cells, the peak components for 1 mmol/L ACh-induced currents were not significantly different as a function of external Ca2+ concentration. However, hump current amplitudes were significantly increased upon removal of external Ca2+. Interestingly, the rate of acute desensitization (inverse of the decay half-time) was significantly increased upon removal of external Ca2+. Thus, although these findings suggest that external Ca2+ at physiological concentrations may slow the rate of α4β2–nAChR desensitization, it still inhibits hump current production.
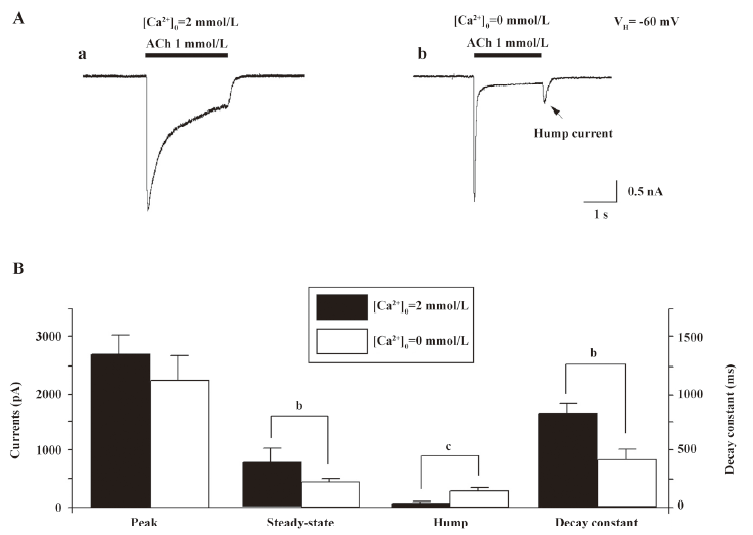
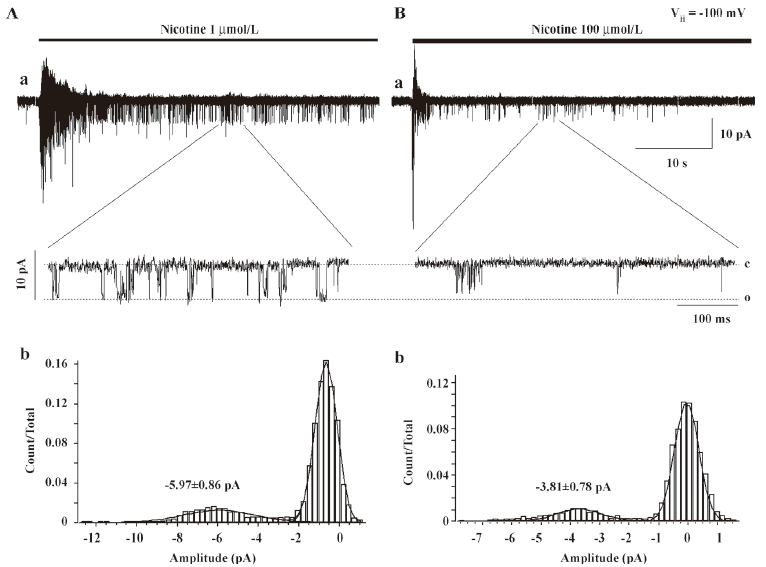
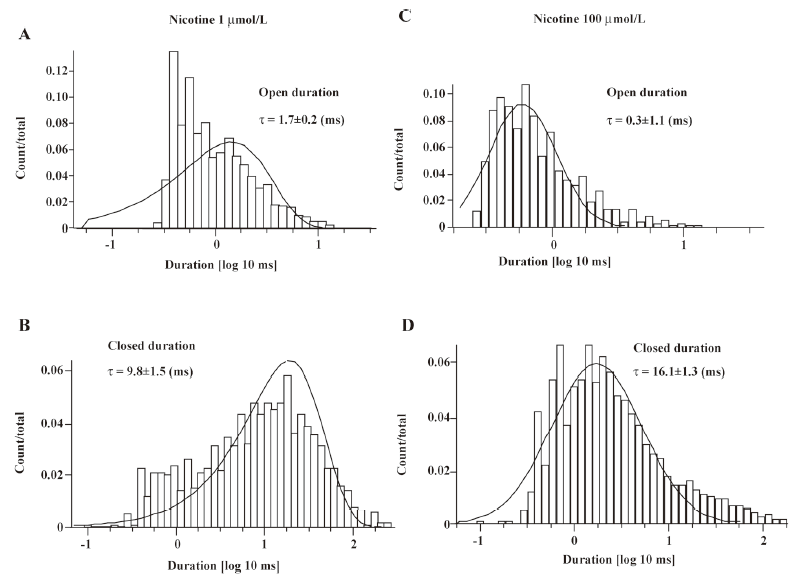
Single-channel evidence for agonist-induced open-channel block of α4β2–nAChR Agonist-induced open-channel block has been studied by single-channel recordings using peripheral muscle-type nAChR[16,27,28]. Key evidence for open-channel block in muscle-type nAChR relies on concentration- and voltage-dependent reductions in mean single-channel currents[27,28]. To determine the single-channel properties of agonist-induced open-channel block, outside-out patch recordings were applied. Figure 9 shows that 1µmol/L nicotine induced inward single-channel currents with dominant amplitude of 5.97±0.86 pA at a VH of –100 mV (Figure 9A). However, as nicotine concentration increased 100-fold, the mean amplitude of single-channel currents was reduced to 3.81±0.78 pA (Figure 9B) at a VH of –100 mV (P<0.05). Figure 9A,9B shows the distribution of the amplitude of single-channel currents induced by 1 or 100 μmol/L nicotine for VH=–100 mV. Figure 10 compares the channel mean open or closed durations for single-channel activities induced by 1 or 100 µmol/L nicotine at a VH of –100 mV. When the cells were exposed to 1 µmol/L nicotine, the mean open and closed times (τ) were 1.7±0.2 and 9.8±1.5 ms (Figure 10A,10B), respectively, whereas exposure to nicotine at 100 µmol/L shortened the mean open time (τ=0.3±1.1 ms; P<0.01; Figure 10C) and prolonged the mean closed time (τ=16.1±1.3 ms; P<0.01; Figure 10D). These results indicate that like muscle-type nAChR[27], transfected human neuronal α4β2–nAChR exhibit the same open-channel block phenomenon represented as concentration- and voltage-dependent features in single-channel activity.
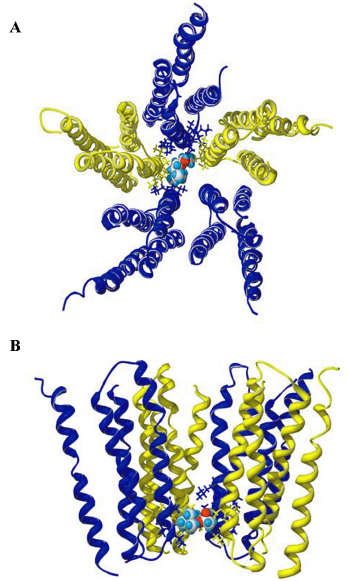
Putative-binding site for ACh in the channel domain of the receptor Using a homology model of the pore-forming transmembrane domains of the α4β2–nAChR and docking software[29] with the selected docking box covering the entire pore region, we successfully docked ACh to a site close to the intracellular end of the pore (Figure 11). This potential ACh-binding site was immediately below the putative channel gate[30]. ACh, with its longitudinal axis oriented horizontally, interacted with residues, mainly at the 6’ and 3’ positions of the second transmembrane domains of 4 subunits (2 α4 and 2 β2). Occupancy by ACh in the narrow part of the pore clearly blocked the ion path of the channel. This location is similar to the binding site for non-competitive antagonists/open-channel blockers (eg picrotoxin) in the pore of the γ-aminobutyric acid type A (GABAA) receptor[31], another member of the cys-loop receptor family. Open-channel blocking behavior and conservation with the binding sites of other channel blockers in the same protein family further support the location of the putative ACh-binding site in the pore of the α4β2–nAChR.
Discussion
Major findings from this study This study demonstrates that the exposure of heterologously-expressed human α4β2–nAChR to select nicotinic agonists at high concentrations induces acute desensitization of functional responses during the period of agonist exposure, but also induces hump currents during drug washout. Magnitudes of peak and hump current responses are directly proportional to whole-cell membrane conductance. Hump currents show voltage dependence and sensitivity to competitive antagonist blockade like those for peak whole-cell current responses mediated via α4β2–nAChR. Prolonged exposure to ACh inducing more pronounced acute desensitization of inward currents also produces decreases in hump current amplitude. Changes in internal ion composition that slow acute desensitization also allow for the maintenance of higher hump current amplitudes, although the removal of external Ca2+ increases both the apparent rate of desensitization and hump current amplitude. Using outside-out patch single-channel recordings, high agonist concentrations reduced current amplitude, shortened the channel mean open time, and prolonged the channel mean closed time, further supporting the agonist-induced open-channel block in human α4β2–nAChR. Collectively, the present evidence suggests the hypothesis that hump currents occur when the agonist producing the blockade of open α4β2–nAChR channels is rapidly released from the channel pore (low-affinity sites) while the agonist is still bound to α4β2–nAChR external-binding sites (high-affinity sites). Both open-channel block and hump current production may occur during synaptic activity that releases a high concentration of ACh, and therefore may underlie some functions of α4β2–nAChR and their modulation.
Other examples of agonist-induced hump currents Hump or rebound currents like those described in this study of human α4β2–nAChR have been found in studies of the effects of high concentrations of ACh on muscle-type α1*–nAChR at frog end-plates[15] and the effects of high (>1 mmol/L) concentrations of pentobarbital on presumed GABAA receptor-gated Cl- currents in isolated frog sensory neurons[32]. In the latter study, the sensitivity of hump currents to the selective GABAA receptor blocker bicuculline was taken as evidence for the reactivation of GABAA receptors by pentobarbital released from blocked channels by rapid washout[32]. Hump currents also were observed in acutely dissociated hippocampal CA1 neurons exhibiting strychnine-induced K+ currents[17] or sevoflurane-induced Cl- currents[18]. More recently, rebound currents mediated by muscle-type nAChR were detected using outside-out patch recordings and modeled using computer simulations consistent with the release from open-channel block[16].
Voltage-dependence, antagonist block, and desensitization of hump currents The very similar current–voltage relationships for peak and hump current production are consistent with both events reflecting the opening of the same kind of channel with the same permeability characteristics. Although a more complete description of the pharmacology for the blockade of hump currents could prove useful, sensitivity to blockade by DHβE was seen for both peak and hump currents or for peak or hump currents individually. Prolonged exposure to ACh or manipulation of intracellular solution composition to enhance the acute desensitization of α4β2–nAChR during agonist exposure also produces desensitization of hump current responses. All of these results are consistent with hump current production by the same channels that mediate peak currents and with reactivation of nAChR channel opening by agonists rapidly leaving the channel after participating in open-channel block.
Concentration-dependence of hump current production by α4β2–nAChR Hump current production by α4β2–nAChR only occurs after washout of higher concentrations of selected agonists. For muscle-type nAChR, it has been hypothesized that the existence of 2 distinct binding sites, one with lower agonist-binding affinity perhaps within the pore and one with higher affinity at the nAChR activation site, may help to explain this effect[27,33]. We entertain a similar explanation for findings in studies of α4β2–nAChR, although it seems that the processes of nAChR inactivation must be more complex than can be explained by a 2-site model. For example, acute desensitization (ie loss of whole-cell inward currents during seconds of exposure to an agonist) of α4β2–nAChR occurs for all nicotinic agonists tested at any concentration that shows significant production of peak whole-cell currents, at least under some conditions of recording (ie using K+ electrodes). However, not all agonists tested were capable of producing hump currents. Thus, the mechanism involved in acute desensitization and the mechanism involved in the loss of nAChR function that is a precursor to hump current production cannot be entirely reconciled. Perhaps a combination of these and other features of agonists are required for hump current production and/or the process that enables it, but the ability to induce hump currents could not be simply attributed to drug action as full or partial agonists, to drug charge or size, or to drug acute potency.
Concentration–response studies indicate that a hump current magnitude produced after exposure to 10 mmol/L ACh is similar to that produced as peak currents by exposure to ~300 µmol/L ACh (ie approximately 35% of the peak current produced by 10 mmol/L ACh). Because hump currents are generally temporally broader than peak currents, perhaps reflecting less synchrony in channel openings than on initial rapid agonist application, the percentage of peak current response may be underestimated. Assume for the moment that the ability of ACh to convert nAChR at rest to the open-channel state is equal to the ability of ACh to convert nAChR from the open-channel blocked state to the open state and that all nAChR are in open-channel block during exposure to 10 mmol/L ACh just before drug washout. The concentration–response studies would then suggest that 35% of open-channel blocked nAChR are reactivated by ACh, leaving channels after 10 mmol/L ACh exposure, meaning that nAChR reactivation would be quite an efficient process, all the more so if the percentage of functionally-available nAChR was reduced due to desensitization. The basis for this efficiency is not presently clear. Outside-out single-channel recordings demonstrated that a higher concentration (100 µmol/L) of nicotine reduced current amplitude, shortened the channel open time, and prolonged the channel closed time, further supporting the hypothesis that open-channel block precedes hump current production.
Practical exploitation of hump currents to illuminate mechanisms of nAChR function Hump currents could serve as important indicators of receptor conditions that might illuminate obscure notions about receptor state during acute desensitization, the existence of open-channel block, and processes of functional recovery from desensitization and open-channel block. There is still disagreement as to whether hump current production occurring during the washout of agonists applied at high concentrations reflects the reactivation of desensitized receptors/channels recovering from open-channel block[32] or rapid recovery from full desensitization[16]. The hypothesis of rapid recovery from full desensitization would seem at odds with the traditional cyclical reaction scheme, in which the recovery of nAChR from full desensitization to the basal state would require more than one step[13,34,35], and with measures showing that such a process requires no less than 1 s[36]. In the latter studies, which were done using a double 0.1 mmol/L ACh pulse protocol to measure the time constant of muscle-type nAChR recovery from receptor desensitization, ~92% desensitization remained 50 ms after the first conditioning pulse[36]. In the present study, the time for washout of agonists was approximately 30 ms, and hump current amplitude was approximately 35% of peak current amplitude, even though a much higher concentration of ACh was used. Thus, either kinetics for recovery from desensitization differs for muscle-type and α4β2–nAChR or hump current production does not reflect recovery from desensitization.
The present findings that hump currents can be desensitized with prolonged agonist exposure or with modulation of intracellular solution also strongly suggest a distinction between desensitization and the process that precedes hump current production. These findings could indicate that loss of nAChR function (whole-cell current decay) during acute exposure to high concentrations of agonists is due to a combination of desensitization and open-channel block. With increased time of agonist exposure, there would be proportionately more desensitization and smaller amplitude hump current production. Decreased internal K+ would decrease the rate and/or extent of whole-cell current decay, increase the proportion of nAChR inactivated via open-channel block, and thereby increase hump current amplitude. From this perspective, perhaps agonist-induced open-channel block protects against desensitization. Recently, Bianchi and Macdonald[37] formulated an “agonist trapping” concept to explain the slow kinetics for the decay of GABAergic inhibitory postsynaptic potentials. They postulated that the conversion of closed or desensitized channel states to open states only occurs if those closed or desensitized states have bound agonists. By analogy, the open-channel block of nAChR may be viewed as a form of agonist trapping, although in this case not at the active site, but through a mechanism that would allow for the rapid release of agonists from the channel pore, leading to apparent recovery from desensitization, manifested as hump current production immediately after agonist washout. However, the present study employing the manipulation of external Ca2+ concentrations suggests that hump current production is more complicated than simple recovery from desensitization and that the extent of open-channel block (revealed by hump current magnitude) may not be simply inversely proportional to rates or extents of desensitization. This is because both hump current amplitude and the rate of desensitization of α4β2–nAChR are decreased in the presence of external 2 mmol/L Ca2+. Although there may be other ways in which Ca2+ modulates nAChR function, perhaps this reflects the existence of different sites on α4β2–nAChR involved in the Ca2+ inhibition of agonist access to open-channel blocking sites and in the Ca2+ inhibition of desensitization.
Physiological significance of hump currents The present study and other previous studies have established evidence for the hypothesis that hump current production indicates the reactivation of receptor function recovering from agonist-induced open-channel block[16–18,32,38]. In studies of nAChR at the zebrafish neuromuscular junction, and consistent with their simulations, Legendre et al[16] found that the synaptic current time-course was influenced by rates of activation and deactivation of nAChR at lower ACh concentrations, but at higher peak concentrations of released ACh the duration of hump (rebound) currents more strongly influenced synaptic current temporal profiles. Thus, at sites proximal to those involved in the release of ACh at high concentrations, open-channel block and subsequent hump current production could help prolong postsynaptic responses and provide distinctive signatures for sites at different distances from nerve terminals that could translate temporal forms into spatial forms of coding critical for synapse formation, maintenance, and remodeling.
Acknowledgements
The contents of this report are solely the responsibility of the authors and do not necessarily represent the views of the aforementioned awarding agencies. The authors thank Dr Ortrud STEINLEIN for the gracious gifts of human α4 and β2 subunit cDNA.
References
- Lindstrom J. Neuronal nicotinic acetylcholine receptors. In: Narahashi T. Editor. Ion Channels. New York:Plenum Press; 1996. p377–450.
- Lukas RJ. Neuronal nicotinic acetylcholine receptors. In: Barrantes FJ. editor. The Nicotinic Acetylcholine Receptor: Current Views and Future Trends, (Neuroscience Intelligence Unit). Austin(TX): Landes Bioscience; 1998. p145–73.
- Lukas RJ, Changeux JP, Le Novère N, Albuquerque EX, Balfour DJ, Berg DK, et al. International Union of Pharmacology. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 1999;51:397-401.
- Jensen AA, Frolund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem 2005;48:4705-45.
- Langley JN. On the contraction of muscle chiesly in relation to the presence of receptive substances. I. J Physiol 1907;36:347-62.
- Gopalakrishnan M, Monteggia LM, Anderson DJ, Molinari EJ, Iattoni-Kaplan M, Donnelly-Roberts D, et al. Stable expression, pharmacologic properties and regulation of the human neuronal nicotinic acetylcholine α4β2 receptor. J Pharm Exper Thera 1996;276:289-97.
- Benowitz NL, Porchet H, Jacob P III. Nicotine dependence and tolerance in man: pharmacokinetic and pharmacodynamic investigations. Prog Brain Res 1989;79:279-87.
- Hsu YN, Amin J, Weiss DS, Wecker L. Sustained nicotine exposure differentially affects a3b2 and α4β2 neuronal nicotine receptors expressed in Xenopus oocytes. J Neurochem 1995;66:667-75.
- Fenster CP, Rains MF, Noerager B, Quick MW, Lester RAJ. Influence of subunit compositions on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci 1997;17:5747-59.
- Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature 1995;374:65-7.
- Cordero-Erausquin M, Marubio LM, Klink R, Changeux JP. Nicotinic receptor function: perspectives from knockout mice. Trends Pharm Sci 2000;21:211-7.
- Court JA, Martin-Ruiz C, Graham A, Perry E. Nicotinic receptors in human brain: topography and pathology. J Chem Neuroanat 2000;20:281-98.
- Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol 1957;138:63-80.
- Ochoa ELM, Li L, McNamee MG. Desensitization of central cholinergic mechanisms and neuroadaptation to nicotine. Molec Neurobiol 1990;4:251-87.
- Adams PR. A study of desensitization using voltage clamp. Pflugers Arch 1975;60:135-44.
- Legendre P, Ali DW, Drapeau P. Recovery from open channel block by acetylcholine during neuromuscular transmission in zebrafish. J Neurosci 2000;20:140-8.
- Ebihara S, Akaike N. Strychnine-induced potassium current in CA1 pyramidal neurons of the rat hippocampus. Br J Pharmacol 1992;106:823-7.
- Wu J, Harata N, Akaike N. Sevoflurane-induced ionic currents in acutely dissociated rat hippocampal CA1 pyramidal neurons of rat. Brain Res 1994;645:303-8.
- Eaton JB, Peng JH, Schroeder KM, George AA, Fryer JD, Krishnan C, et al. Characterization of human α4β2-nicotinic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol Pharm 2003;64:1283-94.
- Lukas RJ, Norman SA, Lucero L. Characterization of nicotinic acetylcholine receptors expressed by cells of the SH-SY5Y human neuroblastoma clonal line. Mol Cell Neurosci 1993;4:1-12.
- Bencherif M, Lukas RJ. Cytochalasin modulation of nicotinic cholinergic receptor expression and muscarinic receptor function in human TE671/RD cells: A possible functional role of the cytoskeleton. J Neurochem 1993;61:852-64.
- Wu J, George AA, Schroeder KM, Xu L, Marxer-Miller S, Lucero L, et al. Electrophysiological, pharmacological, and molecular evidence for alpha7-nicotinic acetylcholine receptors in rat midbrain dopamine neurons. J Pharmacol Exp Ther 2004;311:80-91.
- Wu J, Kuo YP, George AA, Xu L, Hu J, Lukas RJ. beta-Amyloid directly inhibits human alpha4beta2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J Biol Chem 2004;279:37842-51.
- Wu J, Liu Q, Yu K, Hu J, Kuo YP, Segerberg M, et al. Roles of nicotinic acetylcholine receptor beta subunits in function of human alpha4-containing nicotinic receptors. J Physiol 2006;576:103-18.
- Gonzalez-Rubio JM, Garcia de Diego AM, Egea J, Olivares R, Rojo J, Gandia L, et al. Blockade of nicotinic receptors of bovine adrenal chromaffin cells by nanomolar concentrations of atropine. Eur J Pharmacol 2006;535:13-24.
- Huguenard JG, Prince DA. A novel T-type current underlies prolonged Ca2+-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci 1994;12:3804-17.
- Sine SM, Steinbach JH. Agonists block currents through acetylcholine receptor channels. Biophys J 1984;46:277-84.
- Ogden DC, Colquhoun D. Ion channel block by acetylcholine, carbacol and subderyldicholine at the frog neuromuscular junction. Proc R Soc Lond B Bio Sci 1985; 225: 329–55.
- Bursulaya B, Totrov M, Abagyan R, Brooks IIIC. Comparative study of several algorithms for flexible ligand docking. J Comput Aided Mol Des 2004;17:755-63.
- Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature 2003;423:949-55.
- Chen L, Durkin K, Casida J. Structural model for g-aminobutyric acid receptor noncompetitive antagonist binding: Widely diverse structures fit the same site. Proc Natl Acad Sci USA 2006;103:5185-90.
- Akaike N, Mareyama T, Tokutomi N. Kinetic properties of the pentobarbitone-gated chloride current in frog sensory neurons. J Physiol 1987;494:85-98.
- Woodhull AM. Ionic blockage of sodium channels. J Gen Physiol 1973;61:687-708.
- Rang HP, Ritter JM. On the mechanism of desensitization of acetylcholine receptors. Mol Pharmacol 1970;6:357-82.
- Cachelin AB, Colquhoun D. Desensitization of acetylcholine receptor of frog end-plates measured in a vaseline-gap voltage clamp. J Physiol 1989;415:159-88.
- Frank C, Hatt H, Parnas H, Dudel J. Recovery from the rapid desensitization of nicotinic acetylcholine receptor channels on mouse muscle. Neurosci Lett 1992;140:169-72.
- Bianchi MT, Macdonald RL. Agonist trapping by GABAA receptor channels. J Neurosci 2001;21:9083-91.
- Maconochie DJ, Steinbach JH. The channel opening rate of adult and fetal-type mouse muscle nicotinic receptors activated by acetylcholine. J Physiol 1998;506:53-72.