
NK3 and NK4 of HGF enhance filamin production via STAT pathway, but not NK1 and NK2 in human breast cancer cells1
Introduction
It is well documented that cell dissemination is aided by the adequate vascularization of tumors, the release of cells from the primary tumor, and the adhesion of tumor cells to the basement of membrane components prior to the invasion of blood vessels. The disruption of cell–cell junctions by invasive growth is a necessary step for tumor cells to enter directly to the vascular system or indirectly via the lymphatic system. The cells, which are carried to distant parts of the body, extravasate from the vascular system into the parenchyma of target organs[1]. The working mechanism for cancer invasion and metastasis, termed “growth invasion”, has been further studied[2] since it was first proposed[3]. An important feature of the growth invasion mechanism is that the uncontrolled growing cells disrupt cell–cell junctions, thus growth is regarded as the necessary and sufficient condition for cancer invasion.
Cancer has been described as a disease of the cell cycle[4]. The failure of any stage in mitosis and the imbalance of key proteins expressed are some special features of cancer cells. For instance, to complete a cell cycle in human breast cancer cells under the induction of hepatocyte growth factor (HGF) requires a relatively shorter time (6−10 h) compared with the average 20−24 h for normal cells. Therefore, uncontrolled growth is a fundamental feature of cancer cell invasion.
The genetic basis for uncontrolled cell growth and genetic instability for multi-morphogenesis in human breast carcinoma has been studied in cultured cell lines and clinical samples. Tumor survival (eg resistance to living environments), growth (mitogenesis), angiogenesis, metastasis, tubulogenesis, and branching morphogenesis may be induced by growth factors, that is, HGF or epithelial growth factor (EGF) in many cancer cell lines, but all these phenotypes may be more or less associated with 1 or more abnormalities in genetic materials, which are the major internally-driven forces resulting in the uncontrolled growth of tumors, metastatic variants, and cancer progression. Nevertheless, proliferation and efficient metastatic cells can be induced under the stimulation of various extracellular ligands. The invasion properties of cancer can be competitively inhibited by kinase inhibitors or competitive inhibitors of HGF variants (eg NK4, domain from N-terminus to krigle 4)[5].
Apart from the features of uncontrolled growth via a pathway of activation of cyclin D1 in HGF-stimulated pathways in human breast cancer, the disruption of the epithelial monolayer by quickly-growing cancer cells seems to be a necessary stage for invasion[6]. The genetic programs seem to be initiated to bring about the modification of epithelial polarity in overt cytoskeletal rearrangements. Although there is a lack of experimental evidence, some biochemical and structural changes, including integrins in the basal membrane and upregulated expression of metalloproteinases are thought to be required for facilitating cellular invasion[2]. Thus the growth invasion mechanism explains to a certain extent some important features, including that cell proliferation without control tends to invade normal tissues. However, it has been established that cancer cells lose contact inhibition and thus tend to move elsewhere from their primitive site and eventually invade distant tissues, where they survive and grow in an aberrant way and form a secondary colony.
Studies on signal transduction seem to have created notions that the phosphorylation of components in signal transduction pathways is a key chemical step to initiate the cell cycle, or an initiation of the cell cycle has been linked to tumor cell invasion/metastasis as a result of the phosphorylation of components in signal transduction pathways. This concept was challenged by an interesting experiment, in which the treatment of cells with 12-O-tetradecanoylphorbol-13-acetate induced the phosphorylation of Ser-985 of c-Met, a receptor with HGF phosphorylation kinase activity. The phosphorylation was identified to link to protein kinase C (PKC) and protein phosphatase 2A (PP2A)[7], suggesting that not all phosphorylation on c-Met is related to the initiation of the cell cycle. In fact, experimental results have pointed to specific roles of phosphorylation on different components in the HGF–c-Met signaling pathway, that is, tumor survival via the activation of phosphatidylinositol 3-kinase (PI3K)[8]; growth via the activated growth factor receptor-2 (Grb2)/SOS (signaling of screening)/Ras pathway[9-11]; angiogenesis via the STAT (signal transducers and activators of transcription) pathway[12]; tubulogenesis via the recruitment of phospholipase Cγ (PLCγ) to Grb2-associated binder-1 (Gab1)[13]; and branching morphogenesis via the activation of the STAT 3 pathway[12]. Recent studies on signaling components have shown that diverse responses are highly specific and these may include the activation of the mitogen-activated protein kinase pathway[14-17].
Actin is present in all living cells so that an actin control is normally necessary to examine the viability of cells under cultural conditions with chemicals, proteins, submetabolic intermediates, or end products of medical herbs. The formation of actin networks is an important step in cellular morphogenesis, in which filamin and Arp2/3 complex are 2 important links to branch actin filaments, thus they are usually found present in actin networks. A study of filamin A (FLNa)-deficient human melanoma cells showed that FLNa was essential in cells that express it for stabilizing orthogonal actin networks suitable for locomotion[18]. It seems that cells maintain a certain ratio of FLNa and the Arp2/3 complex to actin monomers for cellular locomotion. Further characterization of the 3 components in the formation of acting gelation showed that increasing the activated Arp2/3 complex to the actin ratio raised the FLNa concentration required to induce acting gelation, an effect resulting in actin filament length diminution[19]. This implies that the formation of actin gelation in the presence of FLNa and the Arp2/3 complex stabilizes the actin filaments. The relationship between cellular actin networks and cancer cell invasion seems somehow linked, but their intrinsic relationship has never been reported.
Therefore, it seems that cancer cell metastasis and invasion are the result of a series of complex events involving cellular interactions between targeting cell and basement cell/matrix and signaling transduction in particular pathways. As stated earlier, 2 downstream signaling modulators of c-Met, PI3K and Grb2, were involved in tumor survival or cell growth[13], while phosphoinositide is a downstream effector of the phosphoinositide signaling modulator affecting cell invasion through Matrigel-coated membranes and metastasis in vivo[20]. In the present study, we focused on the molecular mechanism of cell proliferation and invasion, including the phosphorylation of related components in the HGF–c-Met pathway under the stimulation/inhibition of HGF and its variants (NK1, NK2, NK3, and NK4) in human breast cancer cells. The results show that the overexpression of filamins causing imbalanced ratios to actin prohibits cancer cell invasion, but not proliferation, which suggests that cell proliferation and invasion are 2 different events. A possible phosphorylation pathway to regulate the expression of filamins is also analyzed.
Materials and methods
Reagents Taq DNA polymerase, PCR buffer, dNTP mix, and restriction enzymes were purchased from TaKaRa (Tokyo Japan). The intermediate PCR cloning kit (pGEM-T easy vector system) and expression vector pHIS-A were obtained from Promega (Madison, WI, USA). Rabbit antihuman phosphorylated c-Met, mouse antihuman c-Met (Y1234/35) and rabbit antihuman phosphorylated Grb2, mouse antihuman Grb2, rabbit antihuman phosphorylated Gab1, mouse antihuman Gab1, and rabbit antihuman γ or β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Human recombinant HGF was purchased from CytoLab (Rehovot, Israel).
PCR, expression, and purification of recombinant HGF variants The DNA fragments for HGF variants, NK1, NK2, NK3 and NK4, were amplified by PCR using 5’ end forward primer 5’-CGC GGA TCC CAA AGG AAA AGA AGA AAT AC-3’ along with any of the 4 reverse primers: 5’-CGC GGA TCC TAA CAC TGA GGA ATG TCA CAG-3’, 5’-CGC GGA TCC TAG CAT GTT TTA ATT GCA CAG-3’, 5’-CGC GGA TCC TAA CAG TTT GGA ATT TGG GAG C-3’, and 5’-CGC GGA TCC TAA CAA CGA GAA ATA GGG C-3’ to generate DNA fragments for NK1, NK2, NK3, and NK4, respectively. The PCR fragments were cloned into the pGEM-T vector for DNA sequencing and then transferred to pTrcHIS (Invitrogen, San Diego, California, USA) series for expression. Recombinant proteins were purified using Co2+–Sepharose.
Cell maintenance and proliferation assays Breast cancer cells MDA-MB-435S (HTB-129, ATCC, Manassas, VA, USA) were cultured in Leibovitz’s 15 medium (L15) containing 2 mmol/L L-glutamine, 0.01 mg/mL human recombinant insulin (Gibco, Oxford, UK), or 0.5%−10% cosmic calf serum (CCS; Hyclone, Logan, Utah, USA) or 0.5%−10% fetal calf serum in a 37 oC incubator. Cell proliferation assays were carried out as reported[21] with modifications. Briefly, confluent cells were detached in 0.05% trypsin 0.02% EDTA (Biochrom, Berlin, Germany), washed in phosphate-buffered saline (PBS), and diluted in serum-free CCS or fetal bovine serum medium to a final concentration of 1×104 cells/mL. The culture plate was precoated with 10 μg/mL rat tail collagen type I (Upstate, Lake Placid, NY, USA), and 2000 cells were added to each well in in sextuplets . Human native HGF and recombinant HGF variants expressed in Escherichia coli and purified from the Co2+–Sepharose chromatography and dialyzed against PBS, were added to a final concentration of 0.125–8 nmol/L, respectively, and the cells were incubated for 2, 4, and 6 d. The fresh culture medium and ligands were changed every 2 d. At each time point, the cells were stained with 0.2% (w/v) crystal violet in 20% (v/v) methanol for 15 min. The plate was extensively washed with distilled water 3–4 times and allowed to dry. Viable cells stained with the dye were dissolved in 1% (w/v) SDS (0.2 mL/well). The absorbance was recorded at 570 nm in a microplate reader (BioRad 5500, Bio-Rad, Hercules, CA, USA).
Invasion assays Invasion assays were carried out with an 8 μm pore-sized membrane device as reported[22-25]. Briefly, the cells were diluted in L15 to a final concentration of 6.25×105 cells/mL. The cells (0.2 mL) were then added in the upper chambers of 24-well Falcon cell culture inserts (Becton Dickinson, Franklincakes, NJ, USA) precoated with collagen, and the experiment was carried out in sextuplets. HGF and recombinant HGF variants were added respectively in the lower chambers to final concentrations of 2 nmol/L (HGF) and 4 nmol/L (HGF variants). The cells on the apical side of the membrane were removed after 21 h incubation. Cells that migrated to the basal side or well bottom were fixed, stained with crystal violet, and dissolved in 1% (w/v) SDS as described earlier. The absorbance was recorded in a microplate reader at 570 nm.
Immunoprecipitation–Western blot analysis Immunoprecipitation (IP) was initially carried out with the binding of antibodies to specific antigens followed by protein A agarose precipitation. Western blots were carried out to detect phosphorylated proteins or both phosphorylated and non-phosphorylated proteins. Briefly, the cells (2×106 cells) that were detached by 0.05% trypsin were lysed with lysis buffer followed by boiling of the lysate at 100 oC for 5 min. Supernatants that had resulted from centrifugation at 10 000×g for 5 min at 4 oC were saved, and the protein concentration was determined with a protein assay kit (Bio-Rad, USA). A primary antibody (eg rabbit anti-c-Met or rabbit antiphosphorylated Gab1) was added to the lysate, and slow agitation was carried out for 2 h at 4 oC. This was followed by the addition of an equal amount of protein A agarose to bind with the primary antibody. Immunoprecipitants that had resulted from centrifugation at 8500×g for 2 min at 4 oC and dissolved in 50 µL SDS–PAGE buffer were then separated on SDS–PAGE after boiling at 100 oC for 5 min. Protein bands were recognized by mouse antibodies followed by goat antimouse immunoglobulin conjugated with horseradish peroxidase, and the protein bands were revealed with an ECL system (Amersham, Buckinghamshire, UK).
Detection of filamin production by Western blots The cells (1×105) grown in L15 containing 2 nmol/L HGF or 4 nmol/L NK1, NK2, NK3, or NK4 for 3 h were detached in 0.05% trypsin, harvested by centrifugation, and lysed in 200 µL lysis buffer. Supernatants that had resulted from centrifugation were saved, and an equal volume of 2´ SDS–PAGE buffer was added. The samples were heated at 100 °C for 5 min before separation by 10% PAGE. Protein bands were transferred to a nitrocellulose membrane. Western blot detection was accomplished using mouse anti-Gallus gallus actin (amino acid sequence 51−79) and goat antimouse conjugated with horseradish peroxidase, and developed with an ECL system.
Results
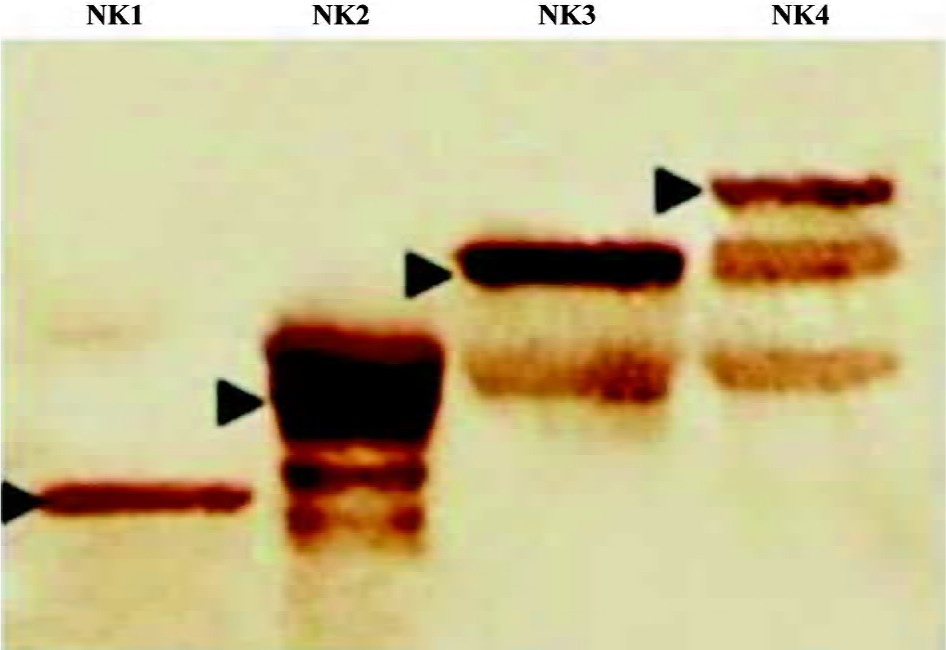
Engineered HGF and expression The human HGF gene was used as a template in protein engineering to amplify DNA fragments for NK1, NK2, NK3, and NK4, respectively, by PCR (Figure 1A). The fragments were cloned into a pGEM-T vector (Figure 1B) for DNA sequencing, which were then cloned in a pTrcHis-A vector for expression (Figure 1C). Western blots showed that all the DNA fragments were successfully expressed in Escherichia coli (Figure 2). NK1 was expressed as a single band, but although the majority of NK2, NK3, and NK4 were expressed as intact mutants, some degraded bands were recognized by the mouse antihuman HGF monoclonal antibody.
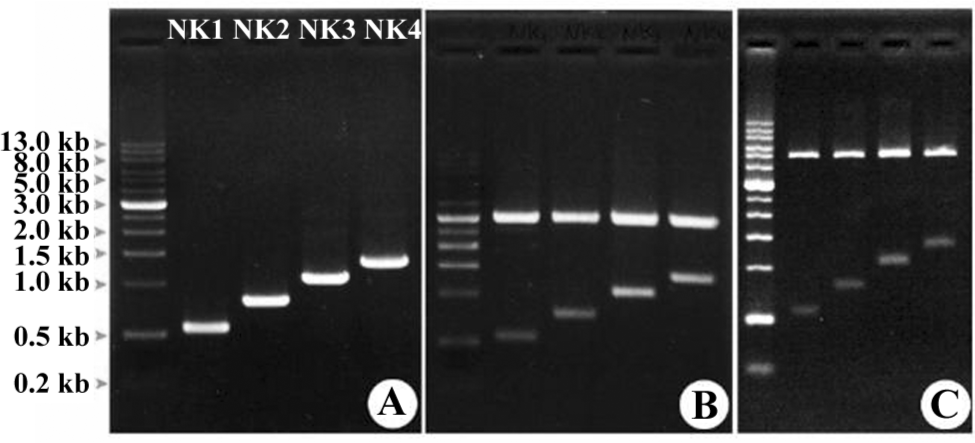
Engineered HGF influence cancer cell proliferation The human breast cancer cell line MDA-MB-435S, which is a model cell line, was used for cellular assays for the proliferation and invasion in the presence of HGF or other growth factors[26,27]. The addition of 1 nmol/L or more recombinant HGF played a significant role in the enhancement of proliferation. Nevertheless, in order to set up the best concentration under our working conditions, we tested various concentrations of HGF and its variants (NK1-NK4) in the assays for proliferation at the range of 0.125−8 nmol/L (Figure 3). The experiment was carried out in sextuplets with growth medium as the control.
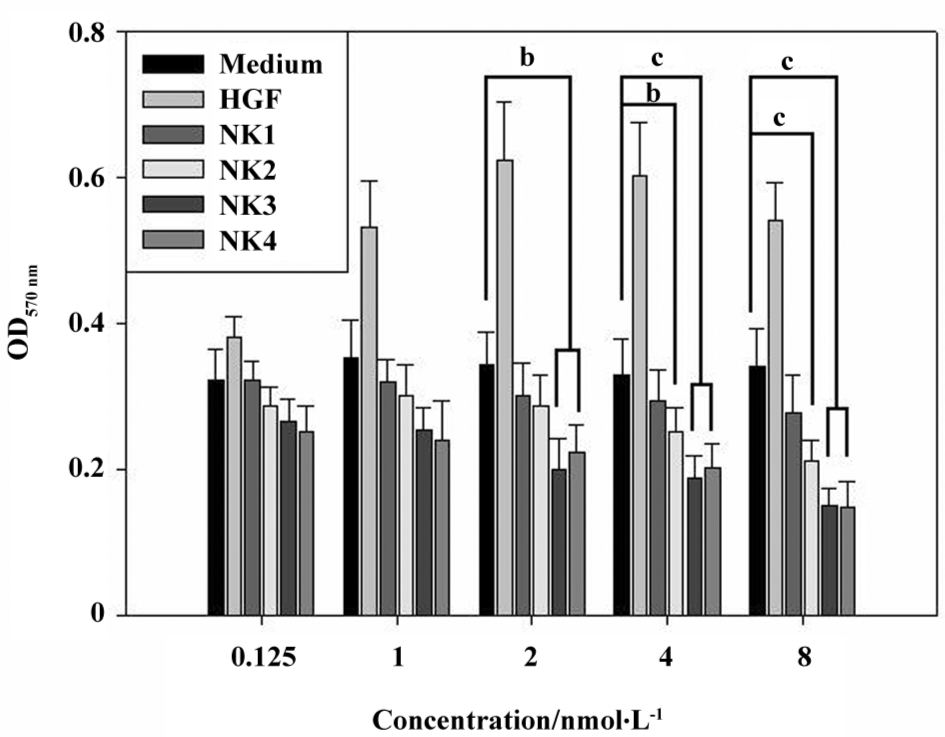
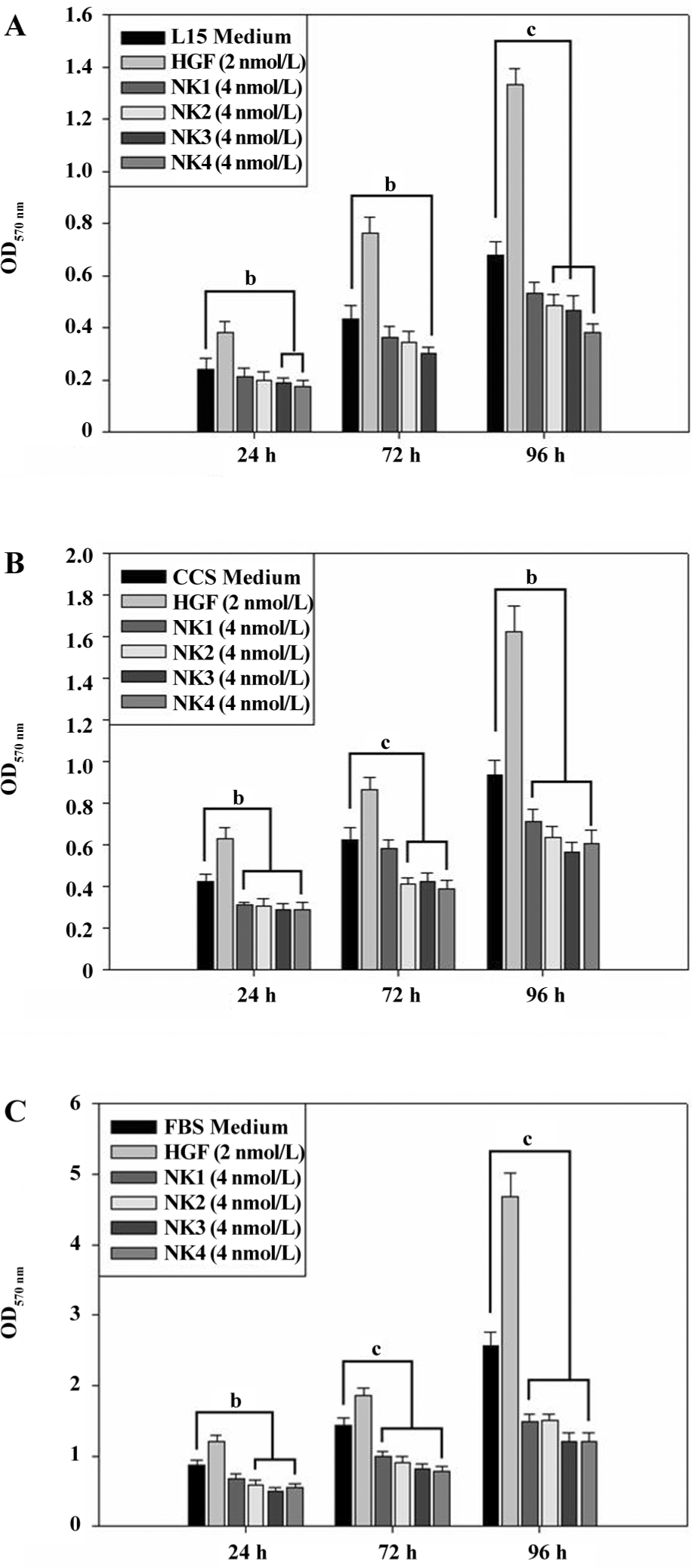
Figure 3 shows that the cells remain at the best viability when the final concentration of additional HGF reaches 1 nmol/L, but cell growth is stopped when the HGF concentration increases to 4 nmol/L. In contrast, HGF variants exhibit a dose-dependent inhibition; higher concentrations showed stronger an inhibition in cell proliferation. NK4/NK3 and NK1/NK2 showed different inhibitory effects on cell proliferation; NK3/NK4 inhibited cell proliferation significantly when the concentration reached 2 nmol/L compared with the medium control, but NK1/NK2 did not. When the concentration of NK2 increased to 4 nmol/L or more, the inhibition of cell proliferation became significant, but NK1 did not; it showed no inhibitory effect even though its concentration increased to 8 nmol/L. Then we assayed the inhibitory effect in different media (L15, CCS, and FBS) at different growing times (24, 72, and 96 h) at a single concentration (2 nmol/L HGF and 4 nmol/L HGF variants). Data are plotted in Figure 4 and show that the proliferation of the cancer cells was well distinguished in the presence of HGF and its variants in L15 (Figure 4A), 1% CCS (Figure 4B), or 1% FBS (Figure 4C) media. The results showed that NK3 and NK4 were rather consistent in the inhibition of cell proliferation in different media, but not NK1. Although the proliferation assays could be carried out in L15 medium, the absorbance showed that the overall cell number was less than that in 1% FBS, suggesting better viability in the FBS medium than L15. Interestingly, cells grown in 1% FBS medium showed significant differences in the presence of different HGF variants at different durations (Figure 4C).
NK3 and NK4 inhibited the invasion of cancer cells Assays for cell invasion were carried out in L15 medium in the presence of 2 nmol/L HGF and 4 nmol/L HGF variants. Cells in the upper chamber invaded through an 8 µm membrane device to the lower chamber in the presence of HGF/HGF variants. After the experiment, the cells in the lower chamber were stained in crystal violet, and the relative cell density was measured.
The results are plotted in Figure 5, and show that cell invasion was significantly inhibited by NK3 or NK4, but not NK1 or NK2 at 4 nmol/L. However, when the concentration of NK1 or NK2 was increased to 20 nmol/L, the inhibition became significant in comparison with the medium control (data not shown).
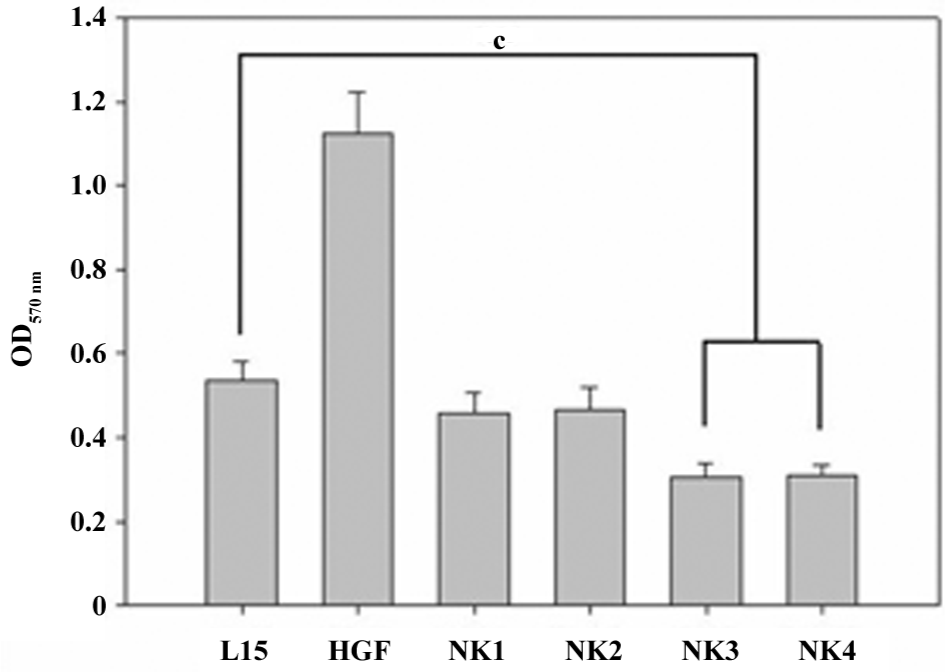
A cross analysis of the effect of all 4 HGF variants on cell invasion showed that all of the recombinant proteins exhibited inhibitory effects on cancer cell invasion either by the inhibition of cell proliferation at 4 nmol/L in 1% FBS (NK1 or NK2) or the inhibition of both proliferation and invasion in all media (NK3 or NK4).
Phosphorylated components under ligand stimulation In order to analyze the possible signaling pathways of recombinant HGF variants, phosphorylated components, including c-Met, Grb2, Gab1, protein kinase B (PKB), and PLCγ were analyzed by IP–Western blots. The results showed that the phosphorylation of c-Met may be stimulated in the presence of HGF, but not HGF variants (NK2–NK4). The results also indicated that NK1 did not seem to inhibit the c-Met phosphorylation. The most obvious difference between recombinant HGF and its variants was the phosphorylation of the cytoplasmic components of PLCγ and Grb2. The stimulation of cells with 2 and 8 nmol/L HGF generated visible phosphorylated PLCγ and Grb2, whereas these components were almost not phosphorylated (PLCγ) or were kept at very low levels of phosphorylation (Grb2) under the stimulation of HGF variants. Gab1 is located between Src and c-Met, but this component remained at a constant phosphorylation level in the cells (Figure 6).
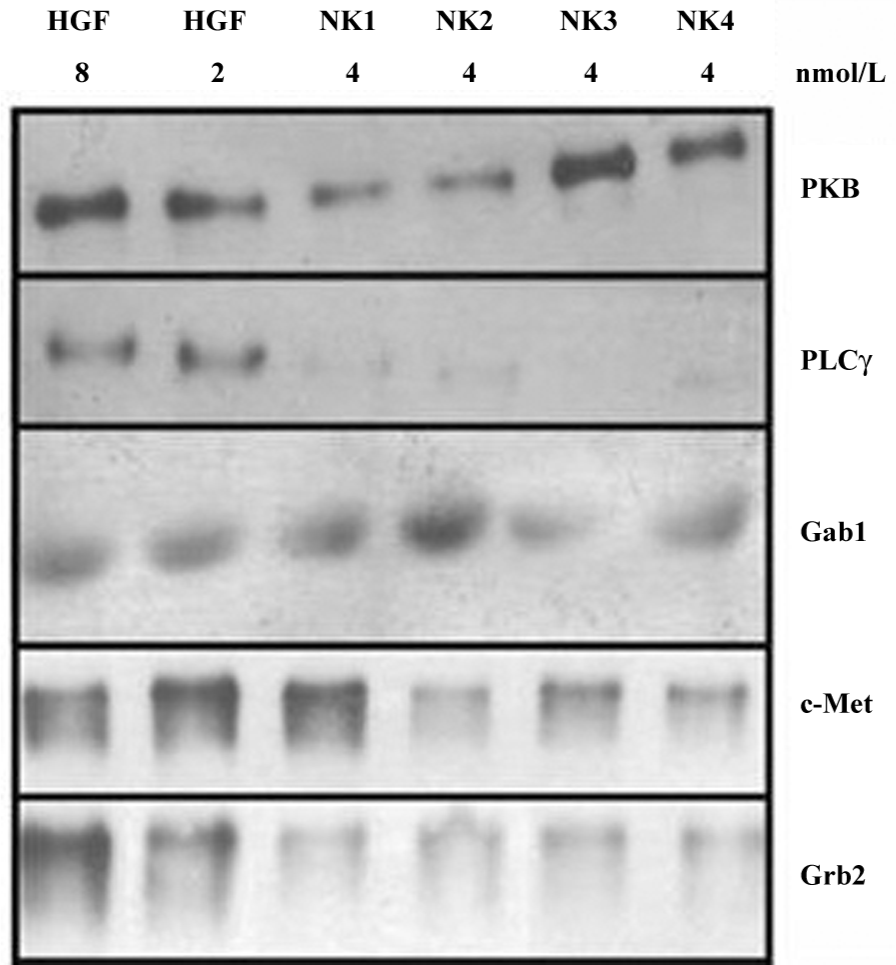
The difference of cytoplasmic component phosphorylation between the HGF variants was also observed. The phosphorylation of Gab1, a component in the c-Met pathway, was enhanced with NK1 and NK2, but not NK3 and NK4. On the contrary, PKB phosphorylation was elevated by NK3 and NK4, but not NK1 and NK2 (Figure 6).
Overexpression of filamins in the presence of NK3 and NK4 Proteins in the actin superfamily in human cells include actins (42, 43, and 48 kDa), filamins (285 kDa filamin A, 230 kDa filamin B, and 165 kDa filamin Bv), FLJ00119 (165 kDa), vimentin (52 kDa), gelsolin (87 kDa), and cdc42 (25 kDa), all of which contain epitopes identical or similar to the sequences of 51 to 70 amino acids of actin from Gallus gallus. The sequence was used to raise antibodies for general purpose. Western blots of total proteins isolated from human breast cancer cells (MDA-MB-435S) incubated with human recombinant or engineered HGF for 3 h showed a striking change in a region of proteins with high molecular weight, including filamins A, B, and Bv (Figure 7).
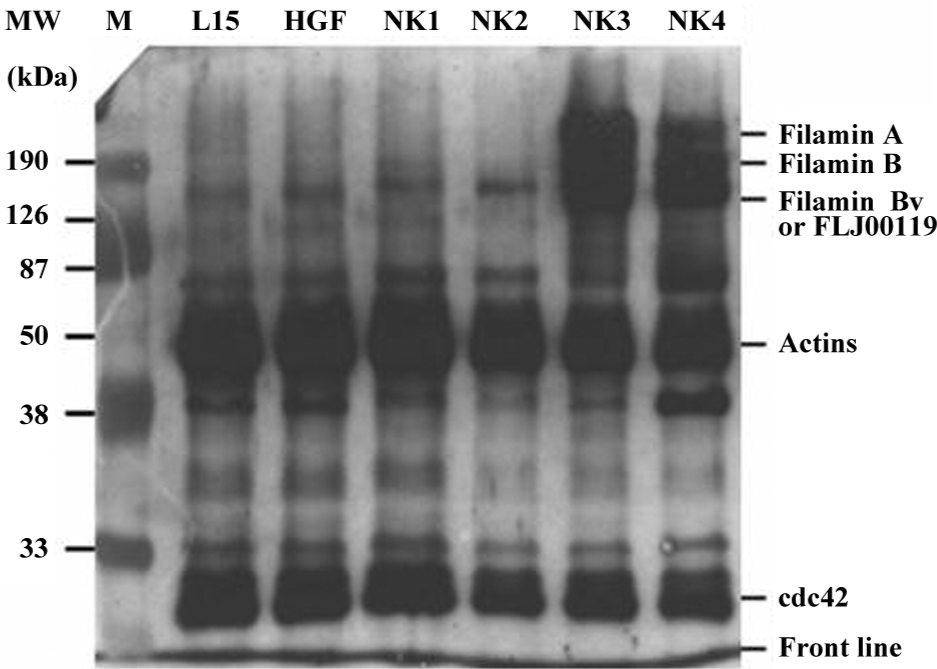
The protein bands from the Western blots were scanned by densitometry and the ratio of filamins to actin in the serum-free control (Figure 7) was approximately 1:40–1:50, which remained unchanged under the stimulation with HGF, NK1, and NK2. However, the ratios of filamin A, filamin B, FLJ00119, and/or filamin Bv to actins were drawn near to 4:1, and there were 10-fold changes when cocultured with NK3 or NK4. The overexpression of filamins and FJL00119 was accumulated to maximum amounts after 3 h of stimulation.
Some unidentified but consistently-occurring protein bands were also recognized by the primary antibodies, but their identities, relations to actins, ligands, or signaling pathways need to be further investigated.
Discussion
Our results clearly demonstrate different signaling transduction pathways in relation to the stimulation of HGF or engineered derivatives in human breast cancer cells. The results suggest that: (1) NK3 and NK4 inhibit the proliferation and invasion of human breast cancer cells via different mechanisms from HGF, NK1, or NK2. That is, signals from HGF, NK1, or NK2 may have similar pathways, but they are different from NK3 or NK4; (2) the inhibition of the proliferation and invasion of cancer cells by HGF variants were exhibited in a dose-dependent manner; and (3) the overexpression of filamins is related to the inhibition of tumor cell migration and invasion.
Proliferation assays in L15 in the presence of HGF or its variants showed that all of the 4 HGF variants caused delayed growth of the cancer cells in a dose-dependent manner, in which NK1 was shown the weakest antagonist in cell proliferation (Figure 3). NK4 had been shown as an antagonist for cancer cell growth in dose-dependent manner[28], and our results showed that NK1 and NK2 were also dose-dependent antagonists. A 7-fold higher concentration of NK1 than NK3 or NK4 was required to exhibit similar inhibitory effects on cell growth. Assays in different media with time course confirmed the inhibitory results of engineered HGF variants (Figure 4). Assays for invasion (Figure 5) in the presence of HGF variants clearly showed that NK1 and NK2 were much weaker antagonists than NK3/NK4. The statistical analysis showed that NK1 and NK2 did not inhibit cell invasion significantly compared with the medium control (Figure 5), whereas NK3 and NK4 were significant inhibitors to the process. This suggests that protein conformations play an important role in binding with receptors and stimulating signal transduction. Moreover, different signaling pathways could be present between NK1/NK2 and NK3/NK4. IP–Western blots for phosphorylated components in the HGF–c-Met pathway (Figure 6) showed that NK3 and NK4 might cause instability of c-Met, Grb2 and PLCγ.
Another new observation in the present work is that both NK3 and NK4 stimulate the production of filamins (Figure 7). Four different subunits were heavily generated after 3 h stimulation with NK3 or NK4, ranging from 165 to 285 kDa. Filamins A, B, and Bv, and FLJ00119 are members of the actin superfamily, and their molecular sizes are located at the region; the lower band (165 kDa) may contain filamin Bv and FLJ00119.
Phosphorylatory routes of proteins or signaling pathways are key chemical steps for initiating or turning off the cell cycle as well as inducing the expression of specific genes, and are therefore involved in various cellular/tissue changes, including mitogenesis, motogenesis, morphogenesis, and angiogenesis. The initiation of the cell cycle thus relates to tumor cell invasion/metastasis as a result of phosphorylation of the components in signaling transduction pathways. Although c-Met phosphorylation has been thought to be the only gate of the HGF/SF (scatter factor)–c-Met pathways to the cell cycle, the phosphorylation identified as a link to PKC and PP2A[7] is not related to the initiation of the cell cycle. Thus we tested some key components including c-Met[8] (the gate of the pathways), which had been shown to be involved in mitogenesis; Grb2, which was related to cell growth via the activated Grb2/SOS/Ras pathway[9-11]; Gab1, which was involved in morphogenesis/tubulogenesis via the recruitment of PLCg to Gab1[13]; and PKB in the STAT pathway[12] to examine their phosphorylation in the presence of HGF and its variants. The results shown in Figure 6 suggest that: (1) HGF increased the total amount of phosphorylated c-Met, PI3K, and Grb2, probably by protecting against the degradation of these components; (2) NK3/NK4 signals were passed via PKB in the STAT pathway, but not NK1/NK2, although all the HGF variants promoted the instability of c-Met, Grb2, and PLC; (3) the production of filamins (Figure 7) may be related to the STAT pathway due to the enhancement of the phosphorylation of PLC, but the signals of NK1 and NK2 did not seem to pass via the same way.
In summary, this paper states clear mechanisms of action of HGF by engineered derivatives: protection against the degradation of signal components (HGF), the promotion of the instability of signal components (all 4 HGF variants), different pathways (eg NK3/NK4 via the STAT pathway, but not NK1/NK2), and enhancement of filamin production (NK3/NK4).
Author contribution
Shi-ping HE designed research; Ya-ling LIN, Hsiu-ling CHEN, Hsiu-maan KUO performed research; Shi-ping HE, Ya-lin LIN analyzed data; Shi-ping HE wrote the paper.
References
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70.
- Gentitle A, Comoglio P. Invasive growth: a genetic program. Int J Dev Biol 2004;48:451-6.
- Welch DR. Tumor progression: analysis of the instability of the metastatic phenotype, sensitivity to radiation and chemotherapy [PhD Thesis]: Houston, TX: University of Texas, 1978.
- North G. Cell cycle: starting and stopping. Nature 1991;351:604-5.
- Jiang WG, Hiscox SE, Parr C, Martin TA, Matsumoto K, Nakamura T, et al. Antagonistic effect of NK4, a novel hepatocyte factor variant, on in vitro angiogenesis of human vascular endothelial cells. Clin Cancer Res 1999;5:3695-703.
- Thiery JP. Epithelial-mesenchymal transitions in tumor progression. Nat Rev Cancer 2002;2:442-54.
- Hashigasako A, Machide M, Nakamura T, Matsumoto K, Nakamura T. Bi-directional regulation of Ser-985 phosphorylation of c-Met via protein kinase C and protein phosphatase 2A involves c-Met activation and cellular responsiveness to hepatocyte growth factor. J Biol Chem 2004; 279: 26 445–52.
- Graziani A, Gramaglia D, Cantley LC, Comoglio PM. The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J Biol Chem 1991; 266: 22 087–90.
- Ponzetto C, Bardelli A, Zhen Z, Maina F. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994;77:261-71.
- Nguyen L, Holgado-Madruga M, Maroun C, Fixman ED, Kamikura D, Fournier T, . Association of the multisubstrate docking protein Gab1 with the hepatocyte growth factor receptor requires a functional Grb2 binding site involving tyrosine 1356. J Biol Chem 1997; 272: 20 811–9.
- Schaeper U, Gehring NH, Fuchs KP, Sachs M, Kempkes B, Birchmeier W. Coupling of Gab1 to c-Met, Grb2 and Shp2 mediates biological responses. J Cell Biol 2000;149:1419-32.
- Boccaccio C, Andò M, Tamagnone L, Bardelli A, Michieli P, Battistini C, et al. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998;391:285-8.
- Gual P, Giordano S, Williams TA, Rocchi S, Van Obberghen E, Comoglio PM. Sustained recruitment of phospholipase C-gamma to Gab1 is required for HGF-induced branching tubulogenesis. Oncogene 2000;19:1509-18.
- Halaban R, Rubin JS, White W. Met and HGF-SF in normal melanocyte and melanoma cells. EXS 1993;65:329-39.
- Chatani Y, Itoh A, Tanaka E. Hepatocyte growth factor rapidly induces the tyrosine phosphorylation of 41-Kda and 43-Kda proteins in mouse keratinocytes. Biochem Biophys Res Commun 1992;185:860-6.
- Gines P, Li X, Brown SE, Nakamura T, Guzelian PS, Heasley E, et al. Inhibitory actions of cyclic adenosine monophosphate and pertussis toxin define two distinct epidermal growth factor-regulated pathways leading to activation of mitogen-activated protein kinase in rat hepatocytes. Hepatology 1996;23:1167-73.
- Adachi T, Nakashima S, Saji S, Nakamura T, Nozawa Y. Mitogen-activated protein kinase activation in hepatocyte growth factor-stimulated rat hepatocytes: involvement of protein tyrosine kinase and protein kinase C. Hepatology 1996;23:1244-53.
- Flanagan LA, Chou J, Falet H, Neujahr R, Hartwig JH, Stossel TP, et al. The Arp2/3 complex, and the morphology and function of cortical actin filaments in human melanoma cells. J Cell Biol 2001;155:511-7.
- Nakamura F, Osborn E, Janmey PA, Stossel TP. Comparison of filamin S-induced cross-linking and Arp2/3 complex-mediated branching on the mechanics of actin filaments. J Biol Chem 2002;277:9148-54.
- Yohem KH, Clothier JL, Montague SL, Geary RJ, Wintersiii AL, Hendrix MJC, et al. Inhibition of tumor cell invasion by verapamil. Pigment Cell Res 1991;4:225-33.
- Wagner JE, Huff JL, Rust WL, Kingsley K, Plopper GE. Perillyl alcohol inhibits breast cell migration without affecting cell adhesion. J Biomed Biotechnol 2002;2:136-40.
- Yu Y, Merlino G. Constitutive c-Met signaling through a nonautocrine mechanism promotes metastasis in a transgenic transplantation model. Cancer Res 2002;62:2951-6.
- Jiang WG, Grimshwa D, Martin TA, Davies G, Parr C, Watkins G, et al. Reduction of stromal fibroblast-induced mammary tumor growth by retroviral ribozyme transgenes to hepatocyte growth factor/scatter factor and its receptor. c-Met. Clin Cancer Res 2003;9:4274-81.
- Sacco MG., Amicone L, Catò EM, Filippini D, Vezzoni P, Tripodi M. Cell-based assay for the detection of chemically induced cellular stress by immortalized untransformed transgenic hepatocytes. BMC Biotechnology 2004;4:5-11.
- Walter-Yohrling J, Cao X, Callahan M, Weber W, Morgenbesser S, Madden SL, et al. Identification of genes expressed in malignant cells that promote invasion. Cancer Res 2003;63:8939-47.
- Parr C, Walkins G, Mansel RE, Jiang WG. The hypatocyte growth regulatory factors in human breast cancer. Clin Cancer Res 2004;10:202-11.
- Kuba K, Matsumoto K, Date K, Shimura H, Tanaka M, Nakamura T. HGF/NK4, a four-kringle antagonist of hepatocyte growth factor, is an angiogenesis inhibitor that suppresses tumor growth and metastasis in mice. Cancer Res 2000;60:6737-43.
- Inoue T, Kataoka H, Goto K, Nagaike K, Igami K, Naka D, et al. Activation of c-Met (hepatocyte growth factor receptor) in human gastric cancer tissue. Cancer Sci 2004;95:803-8.