
Resveratrol improves non-alcoholic fatty liver disease by activating AMP-activated protein kinase1
Introduction
Non-alcoholic fatty liver disease (NAFLD) represents a continuum of hepatic injuries, which progress from simple hepatic steatosis to non-alcoholic steatohepatitis, with some people even ultimately progressing to fibrosis, cirrhosis, and liver failure[1]. The prevalence of NAFLD in the general population is estimated to be between 14% and 24%, and it is so closely associated with obesity, diabetes, and insulin resistance (IR)[1] that it has been proposed that NAFLD be included as a component of metabolic disorders[2,3]. However, the pathogenesis has not yet been clarified and “two hit” hypothesis[4] is now generally accepted. The “two hit” hypothesis believes that IR plays a key role in the development of NAFLD. NAFLD, characterized by hepatic steatosis, can be reversed with early treatment. So far, only weight loss by moderate calorie restriction (CR) has been proven to be an effective treatment for NAFLD[1,5].
Resveratrol (RSV; 3,5,4'-trihydroxystilbene) is a naturally occurring phytoalexin found in red wine. As it possesses diverse biochemical and physiological actions, including estrogenic, anti-inflammatory, and antioxidative properties[6], RSV has received more and more attention[6]. One of the most intriguing findings is that RSV holds multiple beneficial activities mimicking CR[7], such as alleviating obesity-induced IR[8–10] and streptozotocin-induced diabetes in rats[11]. Our previous study showed that CR could greatly improve NAFLD[12]. In addition, early studies[13,14] revealed that RSV might have regulatory effects on lipid metabolism. Therefore, we assumed that RSV could possibly improve NAFLD.
Numerous molecules[6], including silent information regulator 1(SIRT1), nuclear factor-κB, and cyclooxygenase-2, are targeted by RSV, accounting for the multiple biochemical and physiological actions. Most recently, AMP-activated protein kinase (AMPK) was identified as a crucial target of RSV[14,15]. AMPK is a heterotrimeric protein consisting of 1 catalytic subunit (α) and 2 non-catalytic subunits (β and γ). In response to the increase in the cellular AMP:ATP ratio, APMK is physiologically activated by the phosphorylation to the threonine 172 (Thr172) of the α-subunit. Once activated, AMPK phosphorylates its downstream substrates to act as metabolic master switch[16] regulating glucose and lipid metabolism. So the impairment of AMPK activity contributes to IR and ectopic lipid accumulation[17]. Based on this, AMPK cascades have emerged as novel targets for the treatment of metabolic disorders, such as IR and NAFLD[18,19].
In this study, we aimed to investigate whether RSV treatment could improve NAFLD and we explored the possible mechanism. Our results suggest that RSV has the therapeutic potential for preventing or treating NAFLD and IR-related metabolic disorders.
Materials and methods
General reagents RSV was purchased from Upstate Biotechnology (Lake Placid, NY, USA). AMPK-α, phosphorylated AMPK-α (Thr172) antibodies, and β−actin antibody were purchased from Cell Signaling Technology (Beverly, MA, USA). Antirabbit antibodies conjugated to horseradish peroxidase were obtained from Jingmei Biotechnology (Shenzhen, Guangdong, China). TRIzol reagent was purchased from Invitrogen (Carlsbad, CA, USA) and DMSO was from Sigma (St Louis, MO, USA). Insulin was supplied by Novo Nordisk (Copenhagen Baegsvard, Denmark). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from HyClone (Logan, UT, USA) and fetal bovine serum (FBS) was from Gibco (Grand Island, NY, USA). An enhanced chemiluminescence kit and BCA kit was purchased from Pierce (Rockford, IL, USA). The TG kit was from Nanjing Jiancheng Bioengineering Institute (Nanjing, Jiangsu, China). All other reagents were of analytical grade.
Animal protocols and diets Male Wistar rats weighing 180−200 g were obtained from the Experimental and Animal Centre of Tongji Medical College affiliated to Huazhong University of Science Technology (Wuhan, Hubei, China). The animals were housed in individual cages under constant temperature and humidity on a 12-h light/dark cycle. Food and water were provided ad libitum. Protocols for animal experimentation and maintenance were approved by the Animal Ethics Committee at our university and carried out in accordance with the institutional guidelines.
Acute treatment study To determine the AMPK-α phosphorylation level with acute RSV treatment, the rats in the fed state were orally administered with RSV (100 mg/kg) or saline (vehicle; 5 rats in each group). Four hours after administration, the rats were killed.
Chronic treatment study A total of 30 rats were randomly allocated into 2 groups. The animals in the normal control (NC) group (n=10) were fed with standard chow which was purchased from the Experimental Animal Centre of Hubei province (Hubei, China). The rats in the high-fat (HF) feeding group (n=20) were placed on a high-fat diet (59% calorie from lard fat, 21% from protein, 20% from carbohydrate) which was prepared by using lard, sucrose, bile salt, and minerals based on normal chow. Six weeks later, half of the HF group rats were separated into the RSV-treated (HR) group (n=10). The animals in the HR group were orally administered RSV (100 mg·kg-1·d-1), and the HF group was treated with saline. At the end of 16th week, all the animals were killed after they received hyperinsulinemic euglycemic clamp. After the rats were killed, epididymis fat and perirenal fat were isolated and then weighed together as visceral fat mass (VF). The percentage of VF to body weight (BW) was calculated as VF/BW%; liver mass index (LMI) was calculated as the percentage of the liver weight normalized to the BW. Lee’s index[20]of obesity was also computed as follows:
Hyperinsulinemic euglycemic clamp catheterization The modified catheterization technique was used as described previously[21]. Briefly, after overnight fasting, the conscious rats were put into a plastic restraining tube which had a hole to stretch tail out. Tail artery and vein catheterizations were conducted, respectively[21], after the animals had received local anesthesia on the tail root with lidocaine.
Hyperinsulinemic euglycemic clamp The clamp was performed using the protocol previously established[22]. Briefly, the tail vein was used to inject insulin and glucose, while the tail artery was used to obtain blood samples. Insulin infusion (0.25 U·kg-1·h-1) was maintained for 120 min at a constant rate, and a variable infusion of glucose (20% solution) was adjusted to maintain the plasma glucose concentration to the fasting blood glucose level (range±0.5 mmol/L). Blood samples (20 µL) were collected from the tail vein every 5 min for plasma glucose determination. The average glucose infusion rate (GIR; total 12 time points) between the 60th and 120th min at steady euglycemia state (GIR60–120) was used to evaluate insulin sensitivity. The rats were kept quiet and conscious before being killed.
Cell culture and treatment The cultured human hepatoma HepG2 cells grew in DMEM containing normal glucose (5.5 mmol/L glucose) supplemented with 10% FBS, 100 units/mL penicillin, and 100 mg/mL streptomycin as previously described[23]. The cells were incubated in a humidified atmosphere of 5% CO2 at 37 ℃. The HepG2 cells were cultured in complete medium with 10% FBS to 80% cell confluence and subjected to assays after overnight serum starvation. RSV that was dissolved in DMSO was added to the medium while the control group received DMSO only. The final concentration of DMSO did not exceed 0.1%, which did not affect cell viability or AMPK phosphorylation. A cell steatosis model was established by exposing HepG2 cells to a high concentration of glucose (25 mmol/L) and insulin (100 nmol/L) as described previously[24].
Western blot analysis Western blot analysis was performed according to the standard protocol with the following antibodies: AMPK-α and phosphorylated AMPK-α (Thr172) antibodies (both diluted at 1:1000), β-actin antibody (diluted 1:4000), and antirabbit antibodies conjugated to horseradish peroxidase (diluted 1:5000). Briefly, proteins from samples of the liver homogenate or HepG2 cell lysates were subjected to SDS–PAGE. After being transferred onto nitrocellulose difluoride membranes, the proteins were immunoblotted, detected with enhanced chemilumi-nescence, and quantified by densitometry of autoradio-graphs. Phosphorylated AMPK was normalized to the levels of the AMPK-α protein.
RT–PCR Total RNA was isolated using TRIzol reagent according to the manufacturer’s protocol, and 2 µg of each intact total RNA sample was reverse transcribed to complementary DNA. The PCR amplification cycles were carried out using primer sequences presented in Table 1. The PCR products were separated by agarose gel electrophoresis and then visualized by ethidium bromide staining. Specific bands were quantitated by scanning densitometry and normalized to the signal of β-actin.

Full table
Determination of TG accumulation Intracellular triglyceride contents were measured in HepG2 cell lysates and liver homogenates as previously described[14,25]. In brief, 100 mg of liver tissue or approximately 1×107 HepG2 cells was homogenized, respectively. The cell protein concentration was determined by a BCA kit liver homogenates or cell lysates were extracted with a mixture of chloroform and methanol (2:1, v/v). The mixture was vortexed vigorously, allowed to separate into 2 phases, and centrifuged at 655×g for 10 min at 4 °C. An aliquot of the organic phase was evaporated until dry. The isolated lipids were then resuspended in ethanol. Total triglyceride concentrations were then measured by a TG kit according to manufacturer’s instructions. The intracellular triglyceride contents of HepG2 cells were normalized to protein concentrations and expressed as milligrams of lipid per gram of cell protein.
Histopathological examination Formalin-fixed and paraffin-embedded livers were processed routinely for hematoxylin–eosin (HE) staining. Oil-red O staining of frozen liver sections or HepG2 cells on a 6-well plate for lipid deposition detection was performed using an established method[24].
Statistical analysis The statistical analysis was performed with SPSS 13.0 software (SPSS, Chicago, IL, USA). All numeric variables are expressed as mean±SEM. Group statistical comparisons were assessed by one-way ANOVA, and individual comparisons by Student’s–Newmann–Keuls test. A P value of <0.05 was considered statistically significant.
Results
RSV reduced lipid accumulation in the liver and improved liver histology Compared with the NC group, the livers of the HF group were slightly enlarged and had a pale yellow appearance, which was seen both on the capsule and cut surface. The livers of the HR group were almost normal. The livers of the HR group were significantly lighter than that of the HF group, as illustrated by the LMI in Table 2. Next, we observed liver histology by HE staining. The liver sections from the NC group were normal. The livers in the HF group developed obvious ballooning degeneration (Figure 1B). After being treated by RSV, the liver histological changes became almost normalized (Figure 1C). However, no fibrosis was seen in any of the groups. Consistent with this observation, Oil-red O staining of the frozen liver sections showed that high-fat feeding induced overt lipid accumulation (Figure 1E), while RSV considerably decreased the lipid accumulation (Figure 1F). The results were further confirmed by the quantification of TG content in the liver homogenates (Figure 1G).

RSV treatment prevented the development of abdominal obesity and IR As shown in the Table 2, the HR group rats tended to have lower BW, Lee’s index, and VF/BW% compared with the HF group. Lee’s index[20] is a parameter that is similar to the human body mass index for evaluating the obesity of rats. VF/BW% refers to the percentage of visceral fat mass to BW. This suggested that the HF group developed abdominal obesity and RSV seemed to be able to prevent it.

Full table
Hyperinsulinemic euglycemic clamp, which is generally considered as the gold standard to measure in vivo IR, was performed. Compared with the NC group, GIR60–120 significantly decreased while fasting plasma insulin markedly increased in the HF group, suggesting that rats in this group developed hyperinsulinemia and IR (Table 2). However, RSV treatment resulted in elevated GIR60–120 accompanied with reduced level of fasting plasma insulin, indicating that IR in this group was markedly improved.
RSV promoted AMPK phosphorylation both in vitro and in vivo Recent reports showed that RSV could activate AMPK in cultured neuron[15] and HepG2 cells[14]. First, we verified that RSV could activate AMPK in cultured human HepG2 hepatocytes. AMPK phosphorylation levels in Thr172 are known as a marker of AMPK activity. The results showed that RSV treatment resulted in the dose- and time-dependent phosphorylation of AMPK in HepG2 cells by increasing the concentrations of RSV at various times. The phosphorylation level of AMPK was elevated 3.7-fold by RSV stimulation (50 µmol/L) at 24 h (Figure 2A).

To extend the in vitro results, we tested whether treating rats with RSV could stimulate AMPK phosphorylation in the liver. As shown in Figure 2B, compared with the control, the administration of RSV resulted in an increased AMPK phosphorylation level (164% of the control) in the liver within 4 h.
RSV inhibited HepG2 cellular TG accumulation via activating AMPK and downregulating sterol regulatory element binding protein 1c and fatty acid synthase gene expressions As high glucose combined with insulin can greatly induce TG accumulation in liver cells[24], we incubated HepG2 cells with glucose (25 mmol/L) and insulin (100 nmol/L) in the absence or presence of RSV (50 µmol/L) for 24 h. As shown by Oil-red O staining, the incubation led to massive TG accumulation in HepG2 cells (Figure 3), which was largely prevented by RSV (Figure 3). The results were further confirmed by the quantification of the intracellular TG content (Figure 3). In order to know the possible mechanisms behind the lipid-reducing effect of RSV, we detected the phosphorylated AMPK levels in HepG2 cells at the same time. The phosphorylation level of AMPK was reduced to 33.5% of the control (P<0.001) after 24 h incubation with high glucose (25 mmol/L) and insulin (100 nmol/L; Figure 3), but returned to 72.5% of the control under co-incubation with 50 µmol/L RSV (Figure 3).

The transcription factor sterol regulatory element binding proteins 1c (SREBP-1c) and enzyme fatty acid synthase (FAS) have been identified as important targets of AMPK. Upon AMPK activation, the gene expressions of SREBP-1c and FAS are downregulated, ultimately leading to lipid synthesis inhibition[18,26]. So in this study, we further investigated the gene expressions of SREBP-1c and FAS by RT–PCR analysis. SREBP-1c and FAS mRNA increased significantly when the HepG2 cells were incubated with a high concentration of glucose (25 mmol/L) and insulin (100 nmol/L) for 24 h (Figure 3. RSV treatment largely prevented the increase (Figure 3). Taken together, these results implied that RSV treatment resulted in the AMPK activation and downregulation of SREBP-1c and FAS expressions, thus preventing hepatic TG synthesis.
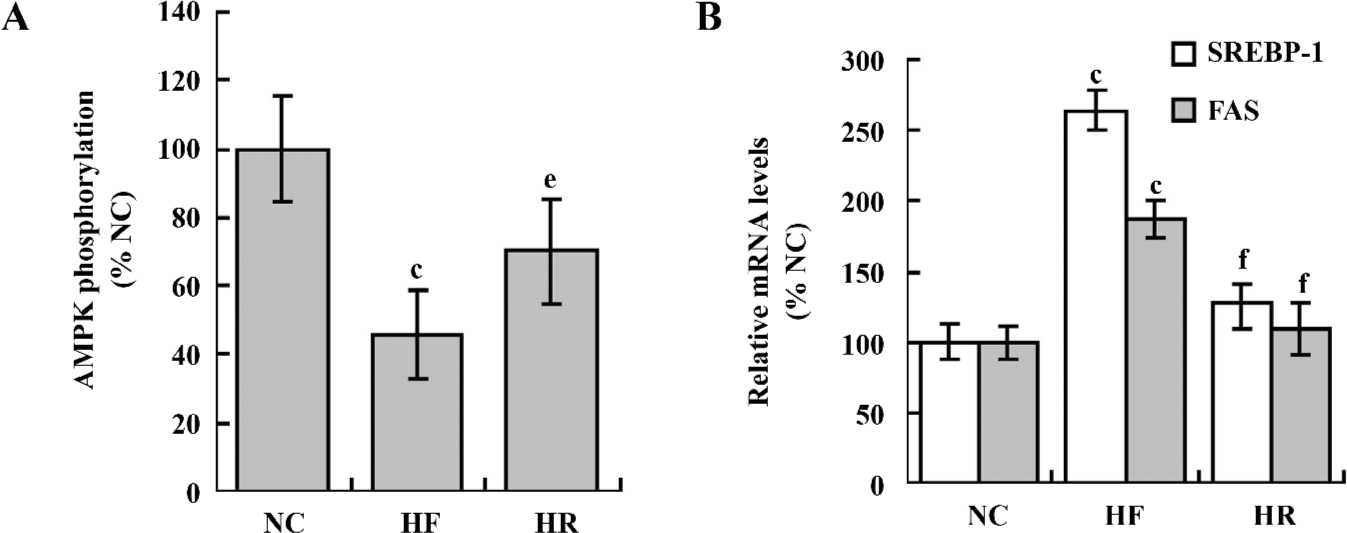
Chronic RSV administration stimulated AMPK phosphorylation and downregulated SREBP-1c and FAS gene expressions in HF feeding rats In addition, we investigated the possible mechanisms behind the beneficial metabolic effects of RSV in vivo. Sixteen weeks of a high-fat diet suppressed the AMPK phosphorylation (Figure 4) and upre-gulated the SREBP-1c and FAS gene expressions (Figure 4). However, RSV treatment induced hepatic AMPK activation (Figure 4). Meanwhile, the gene expressions of SREBP-1c and FAS declined (Figure 4).
Discussion
In the present study, we examined whether RSV could improve NAFLD and we explored the possible mechanism. For this purpose, we first induced the hepatic steatosis in vitro model and NAFLD in vivo model. HepG2 cells that were incubated with a high concentration of glucose and insulin developed steatosis, which was confirmed by Oil-red O staining and quantitation of the total intracellular TG content. Meanwhile, we performed an in vivo study by means of feeding rats a high-fat diet which is a widely-used method of establishing a NAFLD model[27,28]. The liver histology showed that the HF feeding rats had typical steatosis presenting as ballooning degeneration, and numerous lipid droplets. At the same time, the rats fed with HF developed abdominal obesity, as indicated by VF/BW% and Lee’s index. The decreased GIR60–120 and hyperinsulinemia demonstrated that the HF group rats presented IR. All of these observations were in agreement with human NAFLD manifestations, suggesting the successful establishment of the NAFLD model.
NAFLD is characterized by fat accumulation in hepatocytes, visible at light microscopy as small droplets inside the cytoplasm[1]. The pathogenesis of NAFLD has not been well understood until now. Accumulating evidence proves that NAFLD is closely linked to IR[1,3,29], which is often accompanied by abdominal obesity as a result of modern lifestyle changes. The popular “2-hit”[4] hypothesis believes that IR causes fat accumulation in the liver[29]. However, this is disagreement with the study by Varman et al[30] who found that fat accumulation in the liver led to IR based on the fact that a very short timeframe of 3 d for high-fat feeding resulted in specific increases in hepatic fat content without a change of peripheral insulin sensitivity. Considering their findings, we proposed that fat accumulation in the liver and IR caused and potentiated each other creating a vicious cycle of metabolic dysfunction with the resultant development of NAFLD. Reducing TG accumulation as well as improving insulin sensitivity can be an effective therapeutic strategy for NAFLD.
Previous studies have suggested that RSV may have the potential to reduce TG accumulation and improve IR. Early in 1982[13], RSV was found to decrease the rate of hepatic TG synthesis. RSV could also prevent fat accumulation in differentiated adipocytes[31]. It was reported[32] that RSV attenuated alcohol-induced fatty liver pathological changes. Recently, Baur et al[9] and other research groups[8,10] found that RSV improved the insulin sensitivity of mice on a high-calorie diet. However, whether RSV can improve NAFLD has not been determined. In this study, we found that RSV directly prevented TG accumulation in the cell steatosis model. Interestingly, in the animal model of NAFLD, RSV treatment also reduced TG deposition in the liver, improving NAFLD, as confirmed by the pathological observations. IR and abdominal obesity improved correspondingly. In summary, our studies revealed that RSV could improve NAFLD as well as IR, thus hampering NAFLD development.
Next, we explored the molecular mechanism by which RSV improved NAFLD and IR. Recent reports revealed that RSV could strongly activate AMPK in cultured neuron[15] and HepG2 cells[14]. To test if RSV has the same effect in the liver, we first assessed whether RSV could activate AMPK in vitro. Consistency with other studies[14], our data showed that RSV promoted the phosphorylation of AMPK in cultured HepG2 cells in vitro. Furthermore, we observed that 4 h after the administration of RSV, phosphorylated AMPK levels in rat livers were elevated to 164% of the control. AMPK is a metabolic regulator that promotes insulin sensitivity and modulates lipid metabolism[18,26]. Once phosphorylated at Thr172, AMPK is activated and 2 main metabolic effects occur in phosphorylated Thr172, one being the regulation of liver lipogenesis. AMPK suppresses lipogenesis-associated genes, such as SREBP-1c and FAS. SREBP-1c, postulated as a key molecule in the pathogenesis of NAFLD[5], is also an important transcription factor that regulates most of the genes involved in the lipogenesis process, including FAS[18,26]. Activated AMPK abolishes the lipid synthesis process and reduces TG accumulation in the liver. The other metabolic effect is to promote glucose metabolism. Activated AMPK inhibits gluconeogenesis in the liver[33] and enhances glucose uptake in the skeletal muscle[34], contributing to the improvement of IR.
Both in the animal and cell model, we found that AMPK phosphorylation was inhibited. A similar effect was also observed in the hearts of obese Zucker rats and ob/ob mice[17], as well as in the cultured hepatocytes exposed to high concentration of glucose[14]. We found that the gene expressions of SREBP-1c and FAS increased correspondingly along with the decline of the phosphorylation level of AMPK. This result suggested that impaired AMPK activity was responsible for fat accumulation in NAFLD. On the contrary, RSV treatment prevented the decline of phosphorylated AMPK, followed by the downregulation of SREBP-1c and FAS, thus preventing hepatic TG synthesis. Collectively, the beneficial effects of RSV on NAFLD could result from its ability of activating AMPK.
However, it should be noted that RSV has also been identified as an activator of SIRT1[35,36] which is a NAD+-dependent deacetylase. Our previous study[12] revealed that SIRT1 could be involved in the mechanism by which CR improves NAFLD. The activation of SIRT1 by RSV also contributes to the improvement of IR[8–10]. Unfortunately, our study cannot exclude the contribution of SIRT1 to NAFLD, but we suppose that AMPK activation at least in part contributes to the mechanism by which RSV can improve NAFLD. In fact, many of the activities of RSV are similar to the beneficial effects offered by CR, including slowed aging and delaying the onset of chronic disease. The mechanism that RSV mimics CR is complicated and is not fully understood yet. AMPK as well as SIRT1 are some of the enzymes that are activated during CR.
In conclusion, this study indicates that RSV can protect the liver from NAFLD. The activation of AMPK is involved in the mechanism by reducing TG accumulation and improving IR. We believe that the mysterious CR mimetic compound RSV has a promising future in the management of NAFLD and IR-related metabolic disorders. Further research is needed to elucidate this.
References
- Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002;346:1221-31.
- Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 2001;50:1844-50.
- Qureshi K, Abrams GA. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol 2007;13:3540-53.
- Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology 1998;114:842-5.
- Ahmed MH, Byrne CD. Modulation of sterol regulatory element binding proteins (SREBPs) as potential treatments for non-alcoholic fatty liver disease (NAFLD). Drug Discov Today 2007;12:740-7.
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 2006;5:493-506.
- Ingram DK, Zhu M, Mamczarz J, Zou S, Lane MA, Roth GS. Calorie restriction mimetics: an emerging research field. Aging Cell 2006;5:97-108.
- Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab 2007;6:307-19.
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006;444:337-42.
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006;127:1109-22.
- Su HC, Hung LM, Chen JK. Resveratrol, a red wine antioxidant, possesses an insulin-like effect in streptozotocin-induced diabetic rats. Am J Physiol Endocrinol Metab 2006;290:E1339-46.
- Deng XQ, Chen LL, Li NX. The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int 2007;27:708-15.
- Arichi H, Kimura Y, Okuda H, Baba K, Kozawa M, Arichi S. Effects of stilbene components of the roots of Polygonum cuspidatum Sieb. et Zucc. on lipid metabolism. Chem Pharm Bull (Tokyo) 1982;30:1766-70.
- Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes 2006;55:2180-91.
- Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci USA 2007;104:7217-22.
- Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol 1999;277:E1-10.
- Wang MY, Unger RH. Role of PP2C in cardiac lipid accumulation in obese rodents and its prevention by troglitazone. Am J Physiol Endocrinol Metab 2005;288:E216-21.
- Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J Physiol 2006;574:41-53.
- Carling D. The AMP-activated protein kinase cascade—a unifying system for energy control. Trends Biochem Sci 2004;29:18-24.
- Bunyan J, Murrell EA, Shah PP. The induction of obesity in rodents by means of monosodium glutamate. Br J Nutr 1976;35:25-39.
- Guo Z, Zhou L. Dual tail catheters for infusion and sampling in rats as an efficient platform for metabolic experiments. Lab Anim (NY) 2003;32:45-8.
- Kraegen EW, James DE, Bennett SP, Chisholm DJ. In vivo insulin sensitivity in the rat determined by euglycemic clamp. Am J Physiol 1983;245:E1-7.
- Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem 2004; 279: 47 898–905.
- Foretz M, Guichard C, Ferre P, Foufelle F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci USA 1999; 96: 12 737–42.
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957;226:497-509.
- Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 2007;100:328-41.
- Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C. Model of nonalcoholic steatohepatitis. Am J Clin Nutr 2004;79:502-9.
- Svegliati-Baroni G, Candelaresi C, Saccomanno S, Ferretti G, Bachetti T, Marzioni M. A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-alpha and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol 2006;169:846-60.
- Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114:147-52.
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 2004; 279: 32 345–53.
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004;429:771-6.
- Bujanda L, Garcia-Barcina M, Gutierrez-de Juan V, Bidaurrazaga J, de Luco MF, Gutierrez-Stampa M. Effect of resveratrol on alcohol-induced mortality and liver lesions in mice. BMC Gastroenterol 2006;6:35.
- Lochhead PA, Salt IP, Walker KS, Hardie DG, Sutherland C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 2000;49:896-903.
- Hardie DG, Sakamoto K. AMPK. A key sensor of fuel and energy status in skeletal muscle. Physiology (Bethesda) 2006;21:48-60.
- Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem 2005;280:17187-95.
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003;425:191-6.