
PKC promotes proliferation of airway smooth muscle cells by regulating cyclinD1 expression in asthmatic rats1
Introduction
Asthma is characterized by airway chronic inflammation, which is the pathophysiological base of airway hyperres-ponsiveness and remodeling[1,2]. The latter is a complex process that involves all of the component tissues of the airway from the epithelium to the adventitia, which includes epithelial cell damage and mucus gland hypertrophy, reticular basement membrane thickening, alterations in connective tissue composition, and an increase in the content of smooth muscle in the airway wall as a result of hyperplasia and hypertrophy[3]. Increasing evidence indicates that the asthmatic airway smooth muscle (ASM) plays an important role in the remodeling of the airway in asthma[4–6]. The remodeling of the asthmatic rat airway is extensively characterized in vivo; smooth muscle mass is considered to be the main determinant in the airway response to allergen stimulus. McVicker and colleagues concluded that repeat allergen inhalation induced cytoskeletal remodeling in the smooth muscle of rat bronchioles. Increased F-actin content resulted in alterations in the composition and/or arrangement of the contractile apparatus after albumin exposure; such a mechanism may have relevance for airway hyperreponsiveness (AHR) found in allergic asthma[1]. Janssen found that some therapeutic drugs, such as cortisteroids and β2-agonists, were potent inhibitors of ASM cell (ASMC) proliferation, which suggested ASMC was used as a target of asthma therapy[4]. Although the structural changes in morphology concerning ASM in asthma is clear, the mechanism responsible for cellular hyperplasia has not been determined.
Protein kinase C (PKC), a cellular mediator of signal transduction, has also been implicated in controlling a number of mitotic events in mammalian cells[7]. The PKC family consists of at least 11 structurally-related serine/threonine protein kinases and can be divided into 3 subgroups based on differences in their structures and biochemical properties. PKCα, βI, βII, and γ are calcium-dependent and phorbol ester/diacylglycerol-dependent. PKCθ, δ, ε, η, and μ only respond to phorbol ester/diacylglycerol. PKCζ, ι, and λ belong to an atypical PKC subclass that is insensitive to calcium and phorbol ester/diacylglycerol[8]. They are not merely isozymes of identical function, but have specialized roles in cell signaling and specific cellular responses. In recent years we have reported that PKCα contributes to the pathogenesis of asthma by promoting the proliferation of passively-sensitized human ASMC[9]. ASMC are normally quiescent, proliferate at low indices, and remain at the G0/G1 phase of the cell cycle. Studies of bronchial biopsies in mild asthmatics reveal that extracellular stimuli transducer proliferative responses move ASMC through the cell cycle, which comprises of distinct phases G1, S (DNA synthesis), G2, and M (mitosis). D-type cyclins (cyclins D1, D2, and D3), in conjunction with cyclin-dependent kinases (CDK)4 and CDK6, are thought to be key regulators of growth control and G1/S transition[10-12]. The expression of cyclinD1 is upregulated primarily by specific mitogens and is needed for the subsequent expression of cyclins E and A. Mammary gland tissues overexpressing cyclinD1 result in tumor development, which further supports the crucial role of cyclinD1 in regulating cell growth and proliferation[13-15].
However, the results that PKC is involved in the process of ASM remodeling by regulating the expression of cyclinD1 in asthmatic rats have not been reported before. Elucidation of the PKC pathway and its downstream will no doubt lead to novel therapies designed to prevent or reverse these changes, so in this present study, we determined whether PKCα could promote the proliferation of ASMC by regulating the expression of cyclinD1.
Materials and methods
Animal sensitization and allergen exposure Twelve pathogen-free male Brown Norway rats weighing 250-350 g (Experimental Animal Center, Huazhong University of Science and Technology, Wuhan, China) were actively sensitized by an intraperitoneal injection of 1 mg of ovalbumin (OA; Sigma, St Louis, MO, USA) together with 200 μg Al(OH)3 in 0.5 mL of 0.9% NaCl. An 1 mL Bordetella pertussis vaccine containing 6×109 heat-killed organisms was given intraperitoneally as an adjuvant on the first day of the study (d 0). An intraperitoneal injection of 1 mg OA and 200 μg Al(OH)3 in 0.5 mL of 0.9% NaCl was administered on d 7. On d 14–28, the rats were exposed to aerosolized OA 3 times a week[16]. Challenges took place for 30 min in a 0.8-m3 chamber, with free-breathing animals being exposed to 2% (w/v) OA, which was generated by a 402T ultrasonic nebulizer (Siling Instrument, Shanghai, China) with 3 mL/min output. The Animal Care Committee of our institute approved the experimental protocol, and the care and handling of the animals were in accordance with the National Institute of Health guidelines.
Cell culture and groups The ASMC were obtained from asthmatic rats, as previously described[17]. The ASMC were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 1×105 U/L penicillin, and 100 mg/L streptomycin. The cultures were maintained in a humidified atmosphere of 95% air/5% CO2. Third to sixth generation cells were applied in the experiment. The ASMC were cultured in 0.5% serum for 24 h to synchronize at the G0/G1 phase. Subsequently, 10% FBS and DMEM were added to the cultures with or without various treatments for 24 h. The ASMC were divided into 4 groups: (1) Control group, where the cells were cultured in growth medium containing DMSO vehicle after serum starvation; (2) phorbol 12-myristate 13-acetate (PMA; Sigma, USA) group, where the cells were cultured in growth medium containing 10 nmol/L PMA; (3) PMA+2-{1-[3-(amidinothio) propyl]-1H-indol-3-yl}-3-(1-methylindol-3-yl) (Ro31-8220; Sigma, USA) group, where the cells were incubated with 5 μmol/L Ro31-8220 for 1 h before adding 10 nmol/L PMA; and (4) Ro31-8220 group, where the cells were incubated with 5 μmol/L Ro31-8220 only.
Transfection with cyclinD1 recombinant plasmid The constructed expression plasmids of the antisense cyclinD1 (ascyclinD1) gene with the pcDNA3.1 plasmid preserved in our laboratory were digested by BamH1, Xho1, as previous described[18]. Plasmid DNA was extracted using the Endo-free plasmid mini kit (Omega Bio-Tek, lutterworth, Leicestershire UK). Cells were seeded onto a 24-well plate followed by the transient transfection of the cyclinD1 recombinant plasmid. The transfection was performed with Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). Transfected ASMC were randomly divided into 5 groups: (1) pcDNA3.1 group; (2) PMA+pcDNA3.1 group; (3) PMA+pcDNA3.1–ascyclinD1 group; (4) pcDNA3.1–ascyclinD1 group; and (5) Ro31-8220+pcDNA3.1 group.
DNA profile analysis Cells were harvested, washed twice with cold 1× phosphate-buffered saline (PBS), and fixed with 75% ethanol overnight at 4 °C. The samples were subsequently stained with 10 g/L RNase and 50 mg/L propidium iodide and incubated in the dark for 30 min at room temperature. A FACScan machine (Becton Dickinson, Franklin Lakes, NJ, USA) was used to analyze the samples.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay Approximately 5×103 cells were grown in 200 μL 10% FBS-supplemented DMEM medium in 96-well, flat-bottomed plates overnight. After being released from the starvation medium, every group of cells was cultured in the different media 4 times for 20 h according to the group type. At the end-point, the cells were treated with 5 g/L 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma, USA) for 3−4 h. Colorimetric quantification was determined at a 490 nm wavelength by an EL808 ultra micro-plate reader (Bio-Tek Instrument, Winooski, VT, USA) after the addition of DMSO.
Immunocytochemistry staining for proliferating cell nuclear antigen The ASMC were seeded onto 6-well plates at a density of 1×105 cells/mL with covering slides, cultivated, and divided into groups as mentioned before. Immunocytochemistry staining was performed for the expression of proliferating cell nuclear antigen (PCNA; Table 1). The expression of PCNA was performed using a streptavidin/peroxidase kit (Zhongshan Biotek, Beijing, China). The sections were examined light microscopically; the cells positive for PCNA staining were brown in the nuclei. On each slide, 100 cells were randomly counted and then the positive percentage was calculated.

Full table
RT–PCR analysis Total RNA was isolated from the cells using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions. RT was performed with 2 μg RNA using the Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI, USA) according to the manufacturer’s protocol. The PCR amplifications were carried out in the presence of 1.5 mmol/L MgCl2 by using standard PCR buffer, 0.2 mmol/L dNTP, 0.4 mmol/L of each forward and reverse primer, 1 μL RT products, and 0.1 unit Taq DNA polymerase in a total volume of 25 μL. We designed the PCR primers based on GeneBank (Table 2). β-Actin

Full table
Western blot analysis Cells were washed twice with ice-cold PBS, and the whole-cell lysates were obtained using lysis buffer (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 0.02% NaN3, 0.1% SDS, 0.5% sodium deoxycholate1, 1% NP-40, 100 mg/L phenylmethylsulfonyl fluoride, and 1 mg/L aprotinin). The protein content was quantified using Bradford reagent (Bio-Rad, Hercules, CA, USA), and then 100 μg of total protein was subjected to SDS–PAGE on 10% acrylamide gels, followed by transfer to cellulose nitrate membranes (Amersham, Little Chalfont, Buckinghamshire, UK) using a wet transfer unit (Bio-Rad, USA) for 1 h at 200 mA. The membranes were blocked for 1 h with TBS-T (TBS [150 mmol/L NaCl and 25 mmol/L Tris-HCl, pH 7.4] with 0.05% Tween-20) containing 5% (w/v) non-fat dry milk, washed 3 times with TBS-T for 10 min each, and then incubated with the primary antibody (Table 1) at a dilution of 1:500 in TBS-T containing 2% non-fat dry milk overnight at 4 °C, finally with a horseradish peroxidase-conjugated secondary antirabbit antibody (Pierce, Rockford, IL, USA) at a dilution of 1:10000 for 1 h. The bands were developed using an enhanced chemiluminescence detection system (Pierce, USA).
Statistical analysis All data were expressed as mean±SD. F-test was used for the comparison of variance among several groups. The significance of difference between two groups was tested by q-test. The correlations were evaluated using linear regression assay. For all tests, P<0.05 was considered statistically significant.
Results
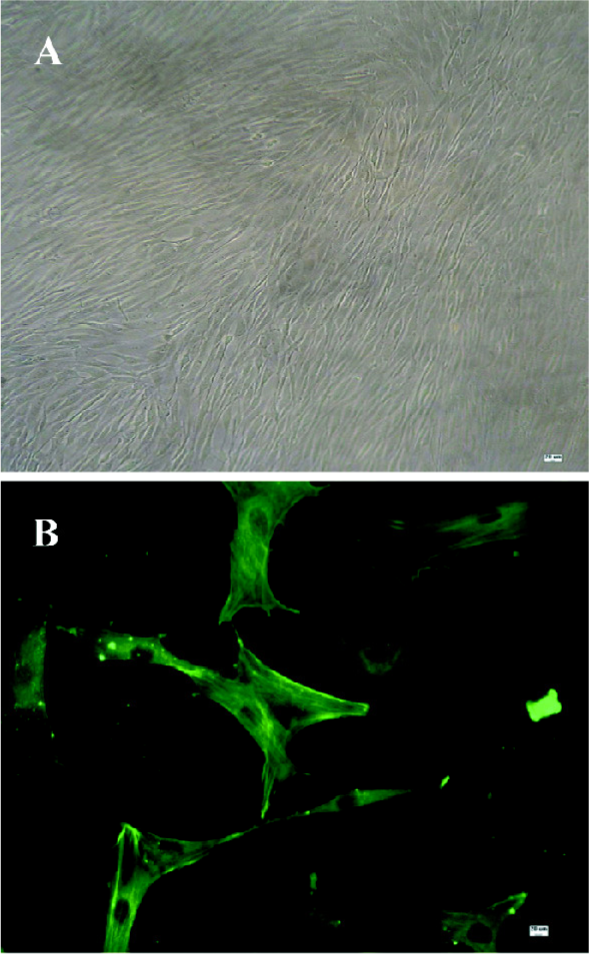
Identification of ASMC The cells were observed by an inverted phase-contrast microscope. They appeared spindle-shaped, having central oval nuclei with prominent nucleoli, as well as the characteristic “hill and valley” appearance (Figure 1A). Confluent monolayers of cells were identified with anti-smooth muscle-actin antibody using an immunofluorescent method to show that the cells were of ASM origin (Figure 1B).
Proliferation levels regulated by PKC in ASMC The activity of PKC has also been indicated to be important in the regulation of cell growth in various types of cells. Furthermore, the asthmatic ASMC were in an active proliferation state. These findings led us to focus on the effect of PKC in the pathophysiology of asthma.
We first investigated the effect of PMA on ASMC proliferation. The cultures were maintained in DMEM containing 10% FBS with or without treatments. They were subjected to MTT assay, a DNA profile analysis, and immunocytochemistry staining for PCNA.
The MTT assay detects mitochondrial dehydrogenase activity and produces the resulting formazan in living cells. Therefore, optical density (OD) represents the living cell numbers. As shown in Figure 2A, PMA increased the OD level markedly, indicating that ASMC proliferation was prompted. The Ro31-8220 treatment reduced the OD value simultaneously.
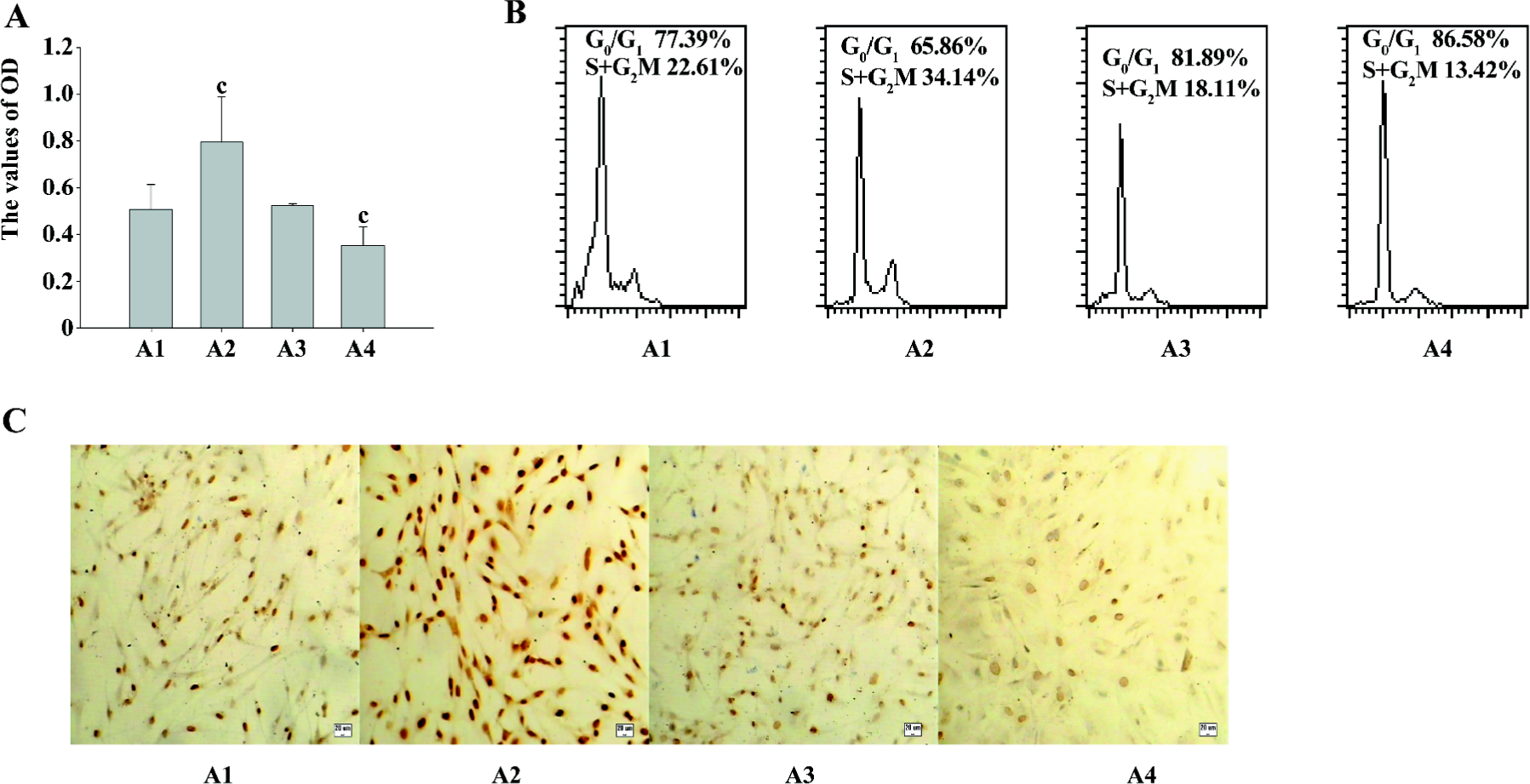
To further examine the growth pattern of ASMC in response to PKC, flow cytometry analysis was conducted. After addition of PMA, the majority of the cells were present at the S+G2M phases in comparison with the untreated control cells, in which a proportion of the cells is still presented at the G0/G1 phase. The opposite effect occurred in the cells treated with Ro31-8220. However, the combination of PMA and Ro31-8220 did not affect the cell cycle progression (Figure 2B), suggesting that the observed growth effects of PMA and Ro31-8220 in ASMC were due to cell cycle progression.
Next, we examined the effect of PMA on the expression of PCNA. The cells were cultured in medium containing PMA or Ro31-8220, alone or in combination for 24 h, and immunocytochemistry staining was performed for the expression of PCNA (Figure 2C). The values of the positive expression rate were 56%±3.6%, 96.9%±7.3%, 52.7%±4.5%, and 42.3%±5.8%, respectively. Collectively, these data indicated that PKC induced the proliferation of asthmatic ASMC.
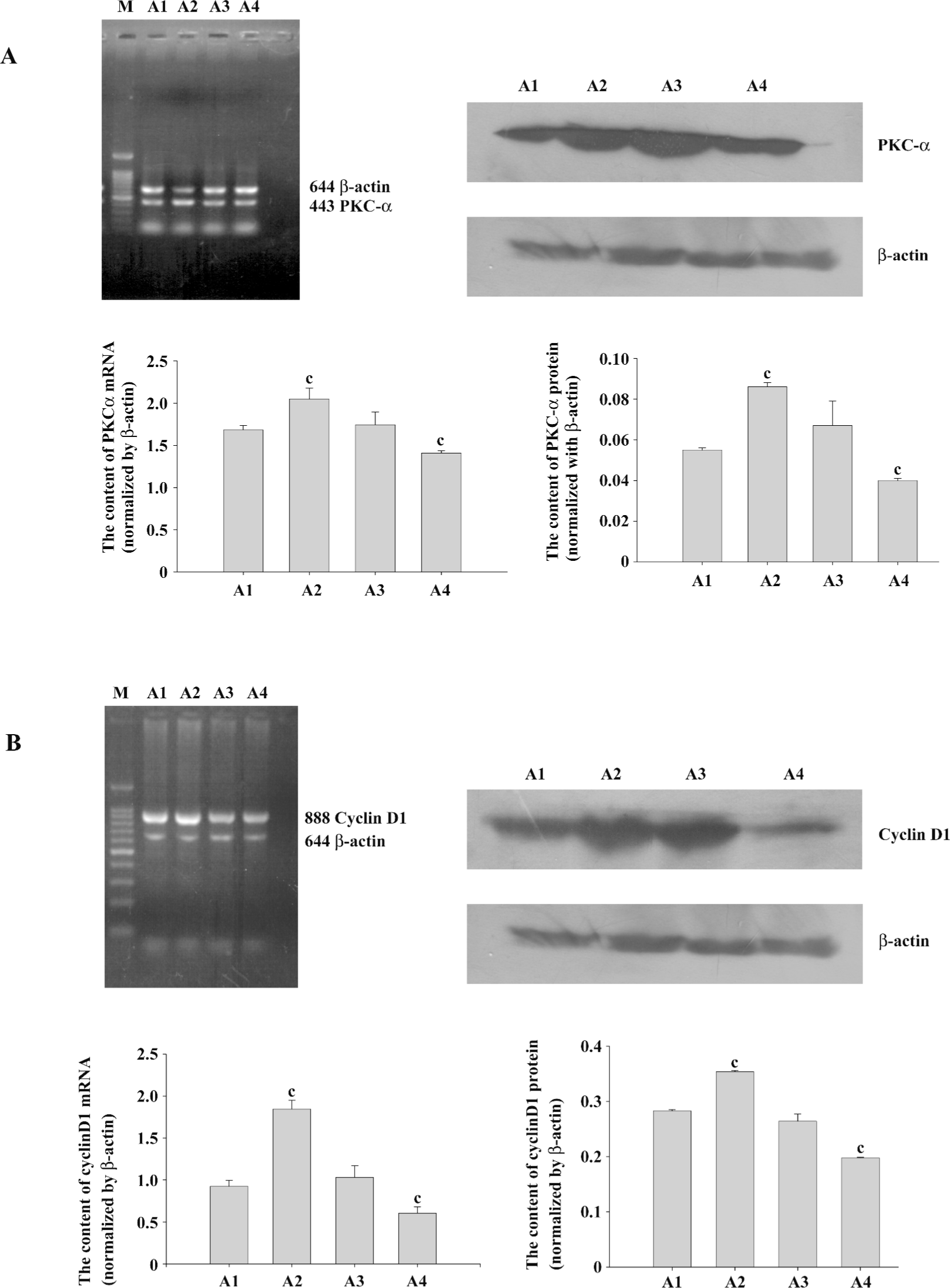
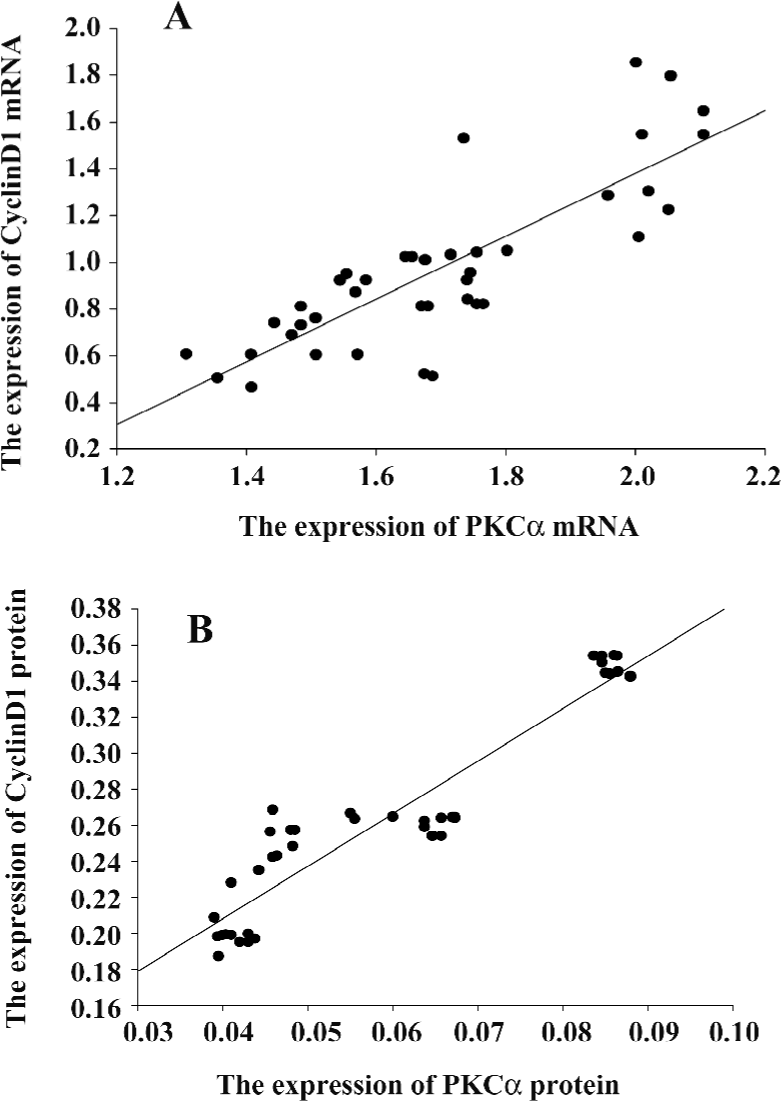
PKCα and cyclinD1 levels stimulated by PMA in ASMC Although the PKC family consists of more than 11 members that participate in diverse cellular responses, it is known that PKCα is the most common isozyme expressed at the protein and mRNA levels in the human lung and airway smooth muscle. To determine whether PMA treatment can induce the activation of PKCα, we examined the mRNA and protein levels. Compared with the control group, the mRNA expression and protein levels of PKCα increased significantly by PMA stimulation and was decreased by Ro31-8220. Treatment with a combination of PMA and Ro31-8220 had no effect on the expression levels of PKCα (Figure 3A). The cyclinD1 mRNA expression level was similar to PKCα (Figure 3B). There was a positive correlation between PKCα and cyclinD1 in the mRNA (r=0.821, P<0.01) and protein(r=0.940, P<0.01) levels (Figure 4). These results suggested that the activation of a conventional PKC, PKCα, probably played an important role in regulating cyclinD1 in asthmatic ASMC.
Proliferation levels regulated by PKC in transfected ASMC It was confirmed that the expression plasmid of ascyclinD1 was successfully recombinated according to the DNA sequence analysis conducted by Invitrogen (Carlsbad, CA, USA).
The MTT results showed that compared with the pcDNA3.1 group, the OD values of cell vitality increased in the PMA+pcDNA3.1 group and decreased in the pcDNA3.1–ascyclinD1 group. Compared with the PMA+pcDNA3.1 group, there was a significant decrease in the cell vitality OD values after PMA+pcDNA3.1–ascyclinD1, pcDNA3.1–ascyclinD1, and Ro31-8220+pcDNA3.1 treatment. There was no difference between the PMA+pcDNA3.1–ascyclinD1, pcDNA3.1–ascyclinD1, and Ro31-8220+pcDNA3.1 groups (Figure 5A).
The flow cytometry results showed that the DNA content was consistent with the MTT assay. The percentage of the S+G2M phases, an index reflecting the proliferation characteristic of cells, were 19.61%, 44.32%, 10.22%, 8.25%, and 6.75%, respectively. PMA+pcDNA3.1 shortened the G1 phase of the cell cycle (55.68%), with an accelerating transition from the G1 phase to the S phase. pcDNA3.1–ascyclinD1 arrested the cell cycle progression at the G0/G1 phase (91.75%) and attenuated the accelerating transition effects induced by PMA (89.78%; Figure 5B), which may explain, at least in part, why the cells being treated with PMA+pcDNA3.1–ascyclinD1 had a lower OD value than the cells transfected with PMA+pcDNA3.1.
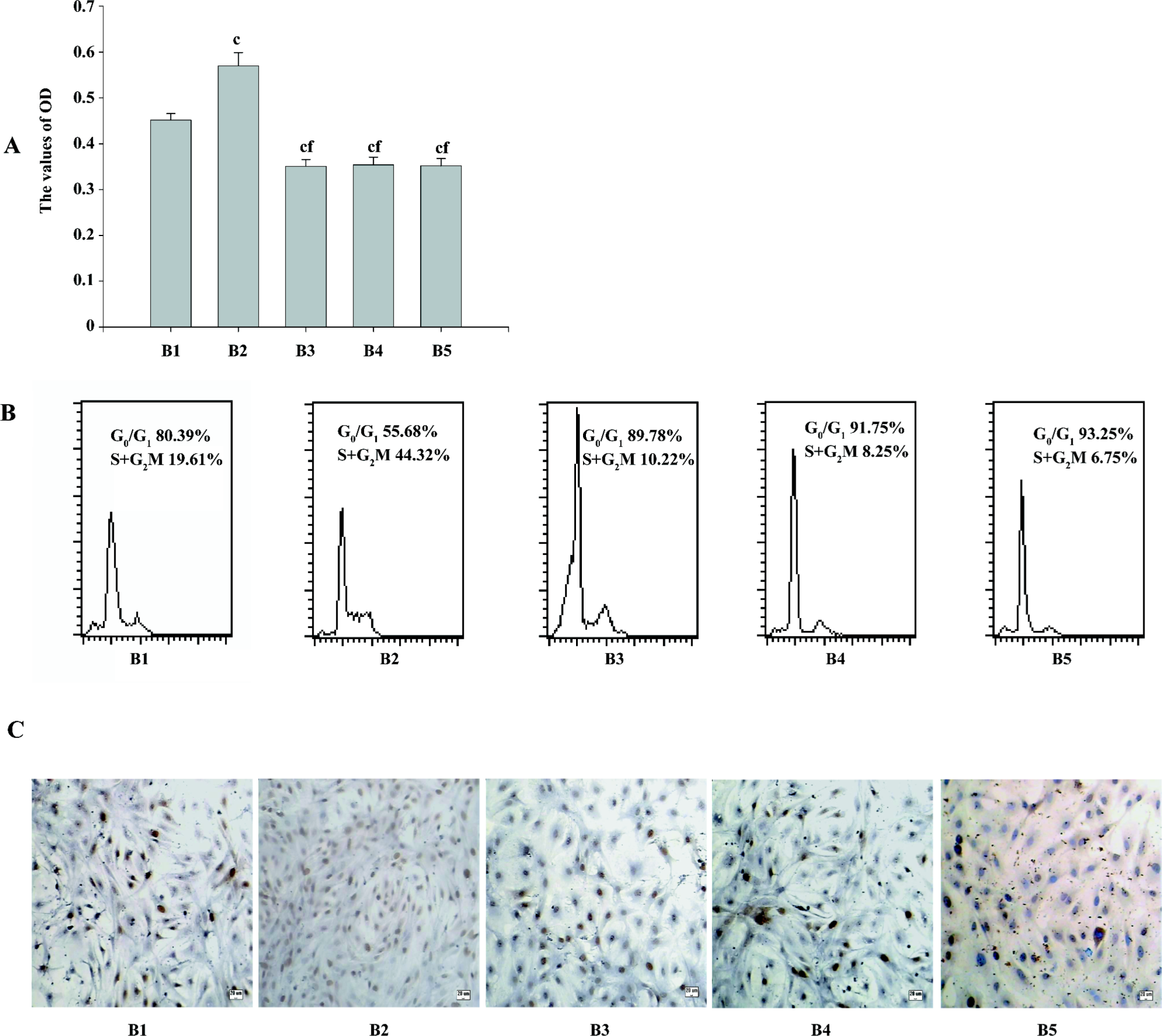
Quantifying PCNA may also indicate the activity of cell proliferation in transfected ASMC. Similar to MTT, the results of the immunocytochemistry staining showed an increase in the positive expression rate of PCNA in the PMA+pcDNA3.1 group and a reduction in the pcDNA3.1–ascyclinD1 group. Compared with the PMA+pcDNA3.1 group, pcDNA3.1–ascyclinD1 also attenuated the effects induced by PMA in quantifying PCNA (Figure 5C).
CyclinD1 expression levels in pcDNA3.1–ascyclinD1-transfected ASMC regulated by PKCα To investigate whether cyclinD1 was involved in the PKC pathway, we constructed an expression plasmid of the ascyclinD1 gene and transfected it to asthmatic ASMC to study the effects of cyclinD1 on the proliferation of cells in asthmatic rats. It was confirmed by restriction endonuclease digestion and the DNA sequence analysis (Invitrogen, Shanghai, China) that the ascyclinD1 plasmid was successfully recombinated.
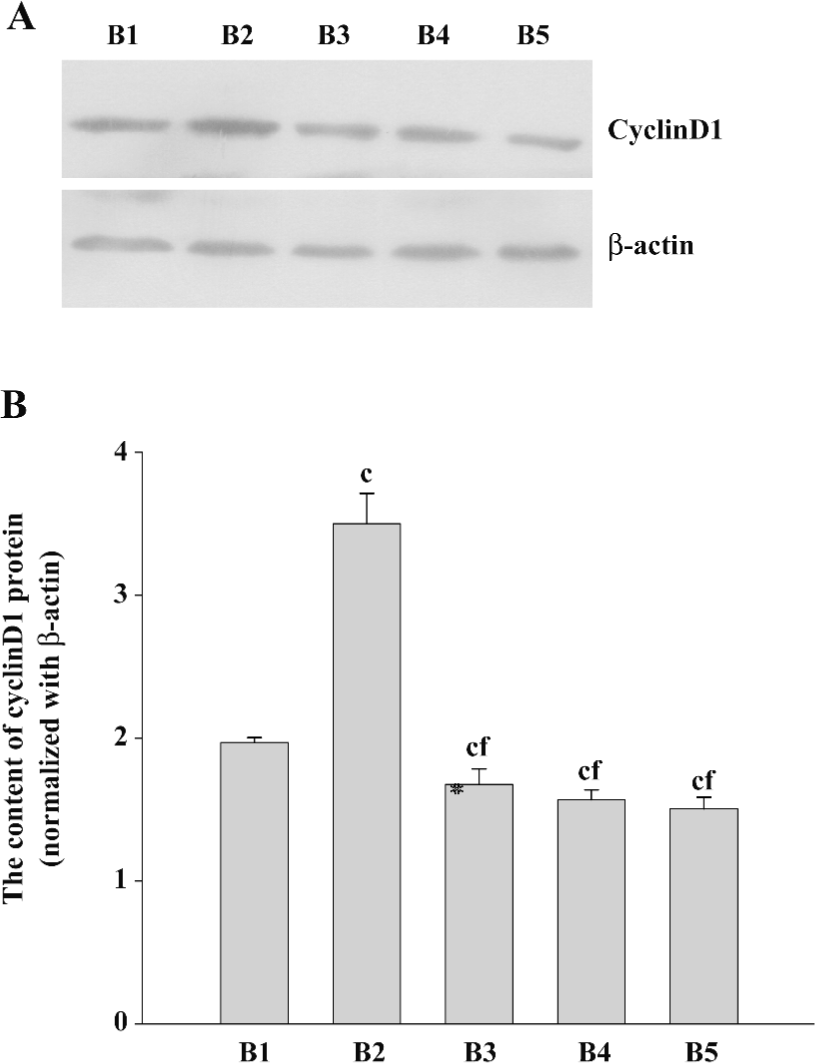
The cyclinD1 expression levels in PMA+pcDNA3.1-transfected cells were higher than that of the pcDNA3.1 group. The expression levels of cyclinD1 in the pcDNA3.1–ascyclinD1-transfected cells were lower than that of the pcDNA3.1 group. Furthermore, pcDNA3.1–ascyclinD1 attenuated the increased cyclinD1 levels induced by PMA as the Ro31-8220+pcDNA3.1 (Figure 6).
Discussion
The specific aim of our study was to observe the effect of PKC rather than the single PKC isoform on the expression of cyclinD1, so PMA, the non-specific agonist for PKC, was chosen as the tool drug. The PKCα isoform was merely selected to support the expression alternation induced by PMA. Our findings showed that PKCα was involved the process of cell proliferation. The increased expression of the cyclinD1 response to activated PKCα may be associated with mechanisms by which PMA promotes the proliferation of ASMC. In addition, pcDNA3.1–ascyclinD1 almost completely attenuated the PMA-induced growth acceleration of ASMC as the Ro31-8220+pcDNA3.1 group, which indicates that the PMA-mediated upregulation of cyclinD1 involves the remodeling of the ASM via the PKC signaling pathway in asthmatic rats.
Although PKC is important for cell proliferation in a number of cell types, little is known about the signaling pathways involved in PKC in ASMC. In the present study, we focused on the involvement of the PKCα isoform in regulating cell growth and revealed whether or not cyclinD1 was at the downstream of PKC pathway in the ASMC of asthmatic rats.
Published observations on the mechanisms of asthma have suggested that there may be a link between the incidence of asthma and various inflammatory mediators, cytokines, and growth factors[19-22]. PKC signaling pathways have been considered as a signaling pathway involved in proliferation, cell cycle progression, differentiation, apoptosis, cytoskeletal remodeling, modulation of ion channels, and secretion initiated by the mitogenic stimulators mentioned earlier[23]. Experiments in vivo and in vitro have suggested their potent activity in the respiratory tracts. In the present study, PMA, a phorbol ester, which is a known substitute for diacylglycerol as a high-affinity ligand for conventional PKC and novel PKC isoforms[24], stimulated PKCα gene expression and protein production in ASMC, and accelerated cell proliferation. On the contrary, Ro31-8220, the selective inhibitor of PKC, repressed the expression of PKCα compared with the control and arrested the cell cycle at the G0/G1 phase, suggesting that increasing PKCα induced by PMA might have contributed to the proliferation of asthmatic ASM. The results were consistent with the article reported earlier, but contrasted with other studies concerning the role of PKC. These divergent observations could at least be partly explained by differences in the subtypes of PKC, the kinds of tissues, and the various concentrations of PMA.
Cyclins have been shown to play an important role in the growth progression of various cells. For example, G1-phase cell cycle proteins stimulate human intestinal smooth muscle cell growth induced by IGF-1[25]. Cyclin-dependent kinase(CDK) inhibited coronary artery smooth muscle cell proliferation and migration[26]. Hammerhead ribozymes targeting against cyclin and E2F1 cooperated to downregu-late coronary smooth muscle proliferation[27]. G1 phase cyclins played a role in the cell cycle progression. The 3 D-type cyclins display significant homology; this has been interpreted as indicating that they may have overlapping functions[28]. However, their expression patterns vary widely among different cell types, suggesting the possibility of distinct functions. CyclinD1 has been the most studied cyclin in ASM biology and has been qualified as a proto-oncogene based on its overexpression in human and animal malignancies as well as its tumorigenic effects in gene transfer experiments with cultured cells and transgenic mice. In bovine ASM, mitogenic stimulation with platelet-derived growth factor (PDGF) induced cyclinD1 transcriptional activation and protein synthesis, consequently resulting in the hyperphosphorylation of phospho-retinoblastoma protein (pRb), which then released the elongation factor E2F that activates DNA polymerase[29]. Consistent with a previous study, the conclusion from our experiment illustrates that cyclinD1 is related to the proliferation of asthmatic ASMC in vitro. The expression of cyclinD1 increased in response to PMA as well as the proliferative activity, but the effect of Ro31-8220 was the opposite, indicating that the PMA-mediated upregulation of cyclinD1 is involved in ASMC proliferation. After being transfected with the pcDNA3.1–ascyclinD1 plasmid containing the Brown Norway rat ascyclinD1 CDS fragment, our data of cell proliferation assessed by MTT, flow cytometry, and the expression rate of PCNA, showed that the ASMC were in a growth suppression state, which further suggested that cyclinD1 could stimulate the proliferation of asthmatic ASMC in vitro.
However, the mechanism of the increased proliferative activity of ASMC induced by PMA is unclear. It should be noted that the upstream of the signaling pathway ultimately regulates the cell cycle through the key proteins of the cell cycle. Another study has also demonstrated that vascular smooth muscle cells exhibit acceleration from the G0 phase to the S phase in the cell cycle progression in response to the activation of PKC[30]. Skaletz-Rorowsk found that the PKC inhibitor can inhibit human and bovine aortic smooth muscle DNA synthesis through the downregulation of basic fibroblast growth factor (bFGF) induced by cyclinD1, yet PMA increased the synthesis of DNA[31]. Accumulating studies have shown that PKC regulates the cell cycle procession by G1-phase cyclins. It is known that phorbol ester-sensitive PKC isoforms, as the cellular receptor for phorbol esters and other tumor promoters, play a significant role in transmitting mitogenic signaling and promoting cell proliferation[32,33]. Thus, the PKC activation-induced cyclinD1 expression further emphasizes the connection between cell cycle regulation and signaling transduction. To the best of our knowledge, the present study is the observation on the expression of cyclinD1 in response to PKC in ASMC. Our results clearly demonstrate that the expression of cyclinD1 is positively relative to the expression of PKCα as expected. ASMC transfected with the pcDNA3.1–ascyclinD1 plasmid simultaneously inhibited PMA-induced S-phase traversal as Ro31-8220+pcDNA3.1, suggesting that PKC signaling pathways lead to the production of cyclinD1 in asthmatic ASMC, and a PKC–cyclinD1 cascade might be responsible for the proliferation in response to PMA. CyclinD1 could be a key downstream target of PKCα; that downstream transcription factor target of PKCα could regulate cyclinD1 transcriptional activity and cell cycle progression, which elucidates the mechanism that PKCα is involved in the remodeling of airway through regulating the cyclinD1 expression.
In conclusion, PKCα stimulates the proliferation of ASMC via the PMA-mediated upregulation of cyclinD1 in asthmatic rats. These findings may provide new insight into the signaling transduction system and the cell cycle regulation in the human respiratory tract, especially in cases of asthma. Further investigations should focus on the different roles of PKC isoforms stimulated by PMA, the cyclins–CDK complexes and CDK inhibitors, and the transduction pathway to determine the pathogenesis of asthma.
Author contributions
Yong-jian XU, Xian-sheng LIU,Jun-gang XIE designed research; Li-fen QIAO, Jin WANG performed research; Chun-ling DU, Jian ZHANG,Wang NI, Shi-xin CHEN contributed new reagents or analytic tools; Li-fen QIAO, Jin WANG analyzed data; Li-fen QIAO wrote the paper.
References
- Mcvicker CG, Leung SY, Kanabar V, Mori LM, Mahn K, Chung KF, et al. Repeated allegen inhalation induces cytoskeletal remodeling in smooth muscle from rat bronchioles. Am J Respir Cell Mol Biol 2007;36:721-7.
- Palmans E, Kips JC, Pauwels RA. Prolonged allergen exposure induces structural airway changes in sensitized rats. Am J Crit Care Med 2000;161:627-35.
- Fixman E D, Stewart A, Martin JG. Basic mechanisms of development of airway structural changes in asthma. Eur Respir J 2007;29:379-89.
- Janssen LJ, Killian K. Airway smooth muscle as a target of asthma therapy: history and new directions. Respir Res 2006;7:123.
- Panettieri RA Jr. Airway smooth muscle: immunomodulatory cells that modulate airway remodeling? Respir Physiol Neurobiol 2003;137:277-93.
- Jeffery PK. Remodeling in asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001;164:S28-38.
- Thompson LJ, Fields AP. Beta II protein kinase C is required for the G2/M phase transition of cell cycle. J Biol Chem 1996; 271: 15 045–53.
- Hofmann J. The potential for isoenzyme-selective modulation of protein kinase. FASEB J 1997;11:649-69.
- Xu SY, Xu YJ, Zhang ZX, Ni W, Chen SX. Contribution of protein kinase C to passively sensitized human airway smooth muscle cells proliferation. Chin Med J 2004;117:30-6.
- Duliæ V, Drullinger LF, Lees E, Reed SI, Stein GH. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inative cyclinE-Cdk2 and cyclinD1-Cdk2 Complexes. Proc Natl Acad Sci USA 1993; 90: 11 034–8.
- Tagliati F, Zatelli MC, Bottoni A, Piccin D, Luchin A, Culler MD, et al. Role of complex cyclinD1/Cdk4 in somatostatin subtype 2 receptor-mediated inhibition of cell proliferation of a medullary thyroid carcinoma cell line in vitro. Endocrinology 2006;147:3535-8.
- Surjit M, Liu B, Chow VT, Lal SK. The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin-cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. J Biol Chem 2006; 281: 10 669–81.
- Roy PG, Thompson AM. Cyclin D1 and breast cancer. Breast 2006;15:718-27.
- Umekita Y, Ohi Y, Sagara Y, Yoshida H. Overexpression of cyclinD1 predicts for poor prognosis in estrogen receptor-negative breast cancer patients. Int J Cancer 2002;98:415-8.
- Chunder N, Mandal S, Roy A, Roychoudhury S, Panda CK. Analysis of different deleted regions in chromosome 11 and their interrelations in early- and late-onset breast tumors: association with cyclin D1 amplification and survival. Mol Pathol 2004;13:172-82.
- Salmon M, Walsh DA, Koto H, Barnes PJ, Chung KF. Repeated allergen exposure of sensitized Brown-Norway rats induces airway cell DNA synthesis and remodelling. Eur Respir J 1999;14:633-41.
- Hirst SJ. Airway smooth muscle cell culture: application to studies of airway wall remodelling and phenotype plasticity in asthma. Eur Respir J 1996;9:808-20.
- Yan KX, Liu BC, Shi XL, You BR, Xu M. Role of cyclinD1 and CDK4 in the carcinogenesis induced by silica. Biomed Environ Sci 2005;18:286-96.
- John M, Au BT, Jose PJ, Lim S, Saunders M, Barnes PJ, et al. Expression and release of interleukin-8 by human airway smooth muscle cells: inhibition by Th-2 cytokines and corticosteroids. Am J Respir Cell Mol Biol 1998;18:84-90.
- Belvisi MG, Saunders M, Yacoub M, Mitchell JA. Expression of cyclo-oxygenase-2 in human airway smooth muscle is associated with profound reductions in cell growth. Br J Pharmacol 1998;125:1102-8.
- Ingram JL, Rice AB, Geisenhoffer K, Madtes DK, Bonner JC. IL-13 and IL-1beta promote lung fibroblast growth through coordinated up-regulation of PDGF-AA and PDGF-Ralpha. FASEB J 2004;18:1132-4.
- Terada M, Kelly EA, Jarjour NN. Increased thrombin activity after allergen challenge: a potential link to airway remodeling? Am J Respir Crit Care Med 2004;169:373-7.
- Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol 2000;279:L429-38.
- Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal 1998;10:529-42.
- Kuemmerle JF, Zhou H, Bowers JG. IGF-I stimulates human intestinal smooth muscle cell growth by regulation of G1 phase cell cycle proteins. Am J Physiol Gastrointest Liver Physiol 2004;286:G412-9.
- Jaschke B, Milz S, Vogeser M, Michaelis C, Vorpahl M, Schomig A, et al. Local cyclin-dependent kinase inhibition by flavopiridol inhibits coronary artery smooth muscle cell proliferation and migration: implications for the applicability on drug-eluting stents to prevent neointima formation following vascular injury. FASEB J 2004;18:1285-7.
- Grassi G, Schneider A, Engel S, Racchi G, Kandolf R, Kuhn A. Hammerhead ribozymes targeted against cyclin E and E2F1 cooperate to down-regulate coronary smooth muscle cell proliferation. J Gene Med 2005;7:1223-34.
- Inaba T, Matsushime H, Valentine M, Roussel MF, Sherr CJ, Look AT. Genomic organization, chromosomal localization, and independent expression of human cyclin D genes. Genomics 1992;13:565-374.
- Möröy T, Geisen C., Cyclin E. Int J Biochem Cell Biol 2004;36:1424-339.
- Liou SF, Yeh JL, Liang JC, Chiu CC, Lin YT, Chen IJ. Inhibition of mitogen-mediated proliferation of rat vascular smooth muscle cells by labedipinedilol-A through PKC and ERK 1/2 pathway. J Cardiovasc Pharmacol 2004;44:539-51.
- Skaletz-Rorowski A, Eschert H, Leng J, Stallmeyer B, Sindermann JR, Pulawski E, et al. PKC delta-induced activation of MAPK pathway is required for bFGF-stimulated proliferation of coronary smooth muscle cells. Cardiovasc Res 2005;67:142-50.
- Wu TT, Hsieh YH, Hsieh YS, Liu JY. Reduction of PKC alpha decreases cell proliferation, migration, and invasion of human malignant hepatocellular carcinoma. J Cell Biochem 2008;103:9-20.
- Dempsey EC, Cool CD, Littler CM. Lung disease and PKCs. Pharmacol Res 2007;55:545-59.