
Hydrogen sulfide regulates lung tissue-oxidized glutathione and total antioxidant capacity in hypoxic pulmonary hypertensive rats1
Introduction
Hypoxic pulmonary hypertension (HPH), an important pathophysiological process in the development of a variety of clinical cardiac and pulmonary diseases[1], has critical influence on the proceeding and prognosis of the diseases. Until recently, the mechanism of HPH has not yet been fully understood. Recent studies have shown that oxidative stress plays a role in the development of pulmonary hypertension[2,3]. Long-term hypoxic animal models have been known to exhibit the overproduction of reactive oxygen species (ROS)[2]. An excess of ROS has been suggested to trigger pulmonary arterial vasoconstriction through the increment of cytosolic calcium in the pulmonary arterial myocyte[4,5]. Hiromitsu et al showed that adrenomedullin, which is not only a potent vasodilator but also an antioxidant, could protect against pulmonary vascular remodeling induced by hypoxia[6]. However, the oxidative/antioxidative state and its mechanism have not been clearly elucidated.
Hydrogen sulfide (H2S) is a newly-found gasotransmitter recently demonstrated to play similar roles as nitric oxide and carbon monoxide in the body, especially in the cardiovascular system[7-11]. Our previous works revealed that the endogenous cystathionine-γ-lyase (CSE)/H2S pathway exited in the vessels[12,13]. As well as this, we found that the endogenous CSE/H2S pathway was down-regulated in hypoxia pulmonary hypertension; exogenously-administered sodium hydrosulfide (NaHS) could reduce pulmonary hypertension and inhibit pulmonary arterial remodeling[14]. Recently, H2S was shown to protect primary rat cortical neurons from oxidative injury by stimulating the synthesis of antioxidant glutathione[15]. Whiteman et al indicated that H2S could significantly inhibit hypochlorous acid-mediated damage to biomolecules and to cultured human neuroblastoma SH-SY 5Y cells[16]. Furthermore Geng et al drew the conclusion that H2S protected the heart from isoproterenol-induced ischemic injury, at least in part, through scavenging oxygen-free radicals and attenuating lipid peroxidation[17]. Thus endogenous H2S might be a new antioxidant in our organism. However, it is unclear whether the endogenous H2S contributes to the pathophysiology of oxidative stress in HPH. We hypothesized that H2S might participate in the regulation of oxidative stress and act as an antioxidant in HPH. To investigate the possible modulating effect of H2S on oxidative stress during HPH and its possible molecular mechanisms, we put rats in a hypoxic environment
Materials and methods
Materials and reagents NaHS was purchased from Sigma (St Louis, MO, USA). TRIzol reagent was bought from Invitrogen (Carlsbad, CA, USA). Oligo d(T)15 primer, dNTP, and M-MLV reverse transcriptase were bought from Promega (San Luis Obispo, CA, USA). All the other chemicals in the study were purchased from Beijing Chemical Reagents (Beijing, China).
Hypoxic pulmonary hypertensive rat model The study was approved by the Animal Research Committee of Peking University (Beijing, China). The rats were exposed to hypoxia according to the methods of Zhang et al[14]. Twenty male Wistar rats (180–220 g) were randomly divided into the control group (n=6), hypoxia group (n=6), and hypoxia+NaHS group (n=8). The rats were exposed to normobaric hypoxia (10% O2) in a transparent plastic hypoxic chamber (for rats in the hypoxia group and the hypoxia+NaHS group) for 3 weeks and 6 h every day. The rats in the control group were housed in identical cages adjacent to the hypoxia chamber and breathed room air. Hypoxia was generated by infusing N2 into the chamber. The degree of hypoxia was maintained by the balance between infusing nitrogen and the inward leak of air through holes in the chamber. For rats in the hypoxia+NaHS group, NaHS, dissolved in physiological saline at a dosage of 14 µmol/kg body weight, was intraperitoneally injected every day before hypoxia. The same volume of physiological saline was injected for the rats in the other 2 groups.
Measurement of hemodynamic parameters and sample preparation Three weeks after hypoxic exposure, the rats were anesthetized with urethane (1 g/kg body weight) intraperitoneally. A silicone catheter (0.9 mm in outer diameter) was introduced into the right jugular vein via a venotomy and passed across the tricuspid valve and right ventricle (RV) into the pulmonary artery. The mean pulmonary artery pressure (mPAP) was simultaneously recorded (90308-11-17-38; SpaceLabs, Issaquah, WA, USA). The mean aortic pressure was also recorded by carotid artery catheterization (90308-11-17-38, SpaceLabs, USA). After the chest was opened, the left lower part of the lung tissues was rapidly removed to liquid nitrogen for quick freezing and then stored at –70 °C. Then the heart was excised and the atria were removed. The free wall of the RV was separated from the heart. These tissues were blotted and weighed using an electronic scale. The wet weight ratio of the RV to left ventricle (LV) plus septum, RV/(LV+septum [SP]), was calculated as an indicator of right ventricular hypertrophy. The blood plasma was also prepared for the measurement of the plasma H2S concentration.
Measurement of H2S concentration in the plasma In a test tube containing 0.5 mL of 1% zinc acetate and 2.5 mL distilled water, 0.1 mL plasma was added. Then 0.5 mL of 20 µmol/L N, N-dimethyl-p- phenylenediamine sulfate in 7.2 mol/L HCl and 0.4 mL of 30 mmol/L FeCl3 in 1.2 mol/L HCl were also added to the test tube for 20 min incubation at room temperature. The protein in the plasma was removed by adding 1 mL of 10% trichloroacetic acid to the solution and centrifuged. The optical absorbance of the resulting solution at 670 nm was measured with a spectrometer (Shimadzu UV 2100, Kyoto, Japan). The H2S concentration in the solution was calculated against the calibration curve of the standard H2S solution.
Measurement of H2S production According to Stipanuk and Beck[18], the lung tissues were homogenized in 50 mmol/L ice-cold potassium phosphate buffer (pH 6.8). Reactions were performed in 25 mL Erlenmeyer flasks. The reaction mixture contained (in mmol/L): 10 L-cysteine 2 pyridoxal 5´-phosphate, 100 potassium phosphate buffer (pH 7.4), and 10% (w/v) homogenates. The total volume of the reaction mixture was 1 mL. A small piece of filter paper was put into the central well of the flask and 0.5 mL of 1% zinc acetate was also added in the central well for trapping evolved H2S in the mixture. The flasks were then flushed with N2 before being sealed with a double layer of parafilm. The catalytic reaction was initiated by transferring the flasks from an ice bath to a 37 °C shaking water bath. After 90 min at 37 °C, the reactions were stopped by injecting 0.5 mL of 50% trichloroacetic acid. The flasks were incubated in the shaking water bath for an additional hour at 37 °C to complete the trapping of H2S. The content of the central well was transferred to test tubes and mixed with 3.5 mL distilled water and 0.5 mL of 20 µmol/L N, N-dimethyl-p- phenylenediamine sulfate in 7.2 mol/L HCl. To each tube, 0.4 mL of 30 mmol/L FeCl3 in 1.2 mol/L HCl was added immediately. After 20 min incubation at room temperature, the optical absorbance of the resulting solution at 670 nm was measured with a spectrometer (Shimadzu UV 2100, Japan). The H2S concentration in the solution was calculated against the calibration curve of the standard H2S solution. For each sample, the measurement was done in duplicate. The H2S production was expressed as nmol·mg–1·min–1.
Measurement of oxidative stress indicators The lower part of the left lung tissues were homogenized in 50 mmol/L ice-cold potassium phosphate buffer (pH 6.8) at the rate of 10% (w/v). The lung homogenates were assayed for total antioxidant capacity (T-AOC), superoxide dismutase (SOD), oxidized glutathione (GSSG), reduced glutathione (GSH), and malondialdehyde (MDA) by using colorimetry.
Measurement of SOD mRNA in lung tissues by quantitative RT–PCR Total RNA was extracted from the rat lung tissues using TRIzol reagent. The cDNA was synthesized using oligo d(T)15 primer and M-MLV reverse transcriptase. The PCR primers were used to amplify the cDNA. The primers of SOD were:
SOD1 (rat-forward): 5'-AAAGGACGGTGTGGCCAAT-3'
SOD1 (rat–reverse): 5'-TCCACCTTTGCCCAAGTCAT-3'
SOD2 (rat–forward): 5'-CGTCACCGAGGAGAAGTACCA-3'
SOD2 (rat–reverse): 5'-GGCTCAGGTTTGTCCAGAAAAT -3'.
The primers were β-actin were:
β-actin (rat–forward): 5'-ATCTGGCACCACACCTTC-3'
β-actin (rat–reverse): 5'-AGCCAGGTCCAGACGCA-3'
Taqman probe was synthesized with 6-carboxyfluore-scein (FAM) reporter dye at the 5'-end and 6-carboxytetramethylrhodamine (TAMRA) quencher at the 3´-end.
The reaction mixture for the real-time PCR (final volume of 50 µL) contained 5 µL 10× PCR buffer, 2 µL of 7.5 µmol/L each primer, 2 µL of 2.5 mmol/L/each dNTP mixture, 0.5 µL of Taq DNA polymerase, and 5 µL of rat lung cDNA. Amplification was performed using an ABI 7300 real-time PCR system (Applied Biosystems, Foster, CA, USA) under the following conditions: 95°C for 5 min, followed by 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. Ten-fold dilution series (1×10-1–1×10-6) were prepared from an extract of the passaged viruses of all genotypes available and analyzed by both PCR methods. Slopes and linear ranges of the standard curves for each genotype were assessed after real-time PCR. Real-time PCR amplified all genotypes with excellent efficiency with correlation coefficients above 0.98.
Measurement of SOD expression in lung tissues by Western blotting Rat lung tissues were homogenized and lysed. Equal amounts of protein were boiled and separated by SDS–PAGE and electrophoretically transferred to a nitrocellulose membrane according to experiment protocols. The primary antibody dilution was 1:8000 for the SOD1 (Stressgen, Ann Arbor, MI, USA) and SOD2 antibodies (Stressgen, USA) and 1:10000 for β-actin (Labvision, Fremont, CA, USA). Horseradish peroxidase-conjugated secondary antibody (Santa Cruz, CA, USA) was used at 1:12000. The immunoreactions were visualized by electrochemiluminescence (ECL) and exposed to X-ray film (Kodak Scientific Imaging Film, Rochester, NY, USA).
Data analysis All data were expressed as mean±SD. For the comparison of differences among the 3 groups, one-way ANOVA followed by a post-hoc analysis (listed significant difference test, LSD) was conducted using SPSS 13.0 statistic analysis software (SPSS, Chicago, IL, USA). To compare the changes of the 2 groups, we selected the independent-samples t-test method. A value of P<0.05 was considered statistically significant.
Results
Hemodynamics After 3 weeks of hypoxia, the mPAP was increased by 45.3% compared with rats in normoxic environment (23.7±2.2 vs 16.3±3.7 mmHg, P<0.01) in the present study. The ratio of RV/(LV+SP) was also increased by 41.4% in the rats of the hypoxia group compared with that of the control group (P<0.01). These results showed that the HPH rat model was reproduced successfully. To examine if the exogenous H2S impacts on hypoxic pulmonary hypertension, we administered 14 µmol·kg–1·d–1 NaHS, a donor of H2S. The results showed that the mPAP of the hypoxia group was markedly decreased, and the RV/(LV+SP) was also reduced (all P<0.01), as shown in Table 1 and Figure 1.
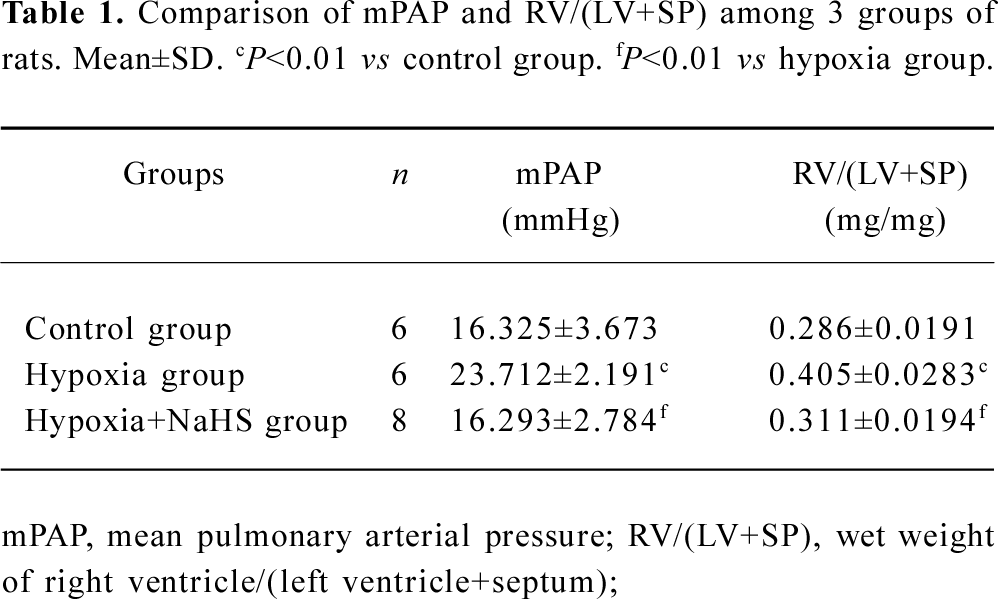
Full table
Plasma H2S concentration and H2S production of lung tissues The plasma level of H2S and its production in the lung tissues decreased significantly in the hypoxia group compared with the control group (187.2±13.1 vs 299.6±12.4 µmol/L, 0.138±0.013 vs 0.289±0.036, respectively, all P<0.01). After treatment with NaHS, the plasma level of H2S and H2S production in the lung tissues increased to 1.7- and 2.3-fold, respectively, in the hypoxia+NaHS group compared with the hypoxia group, as shown in Table 2 and Figure 2.
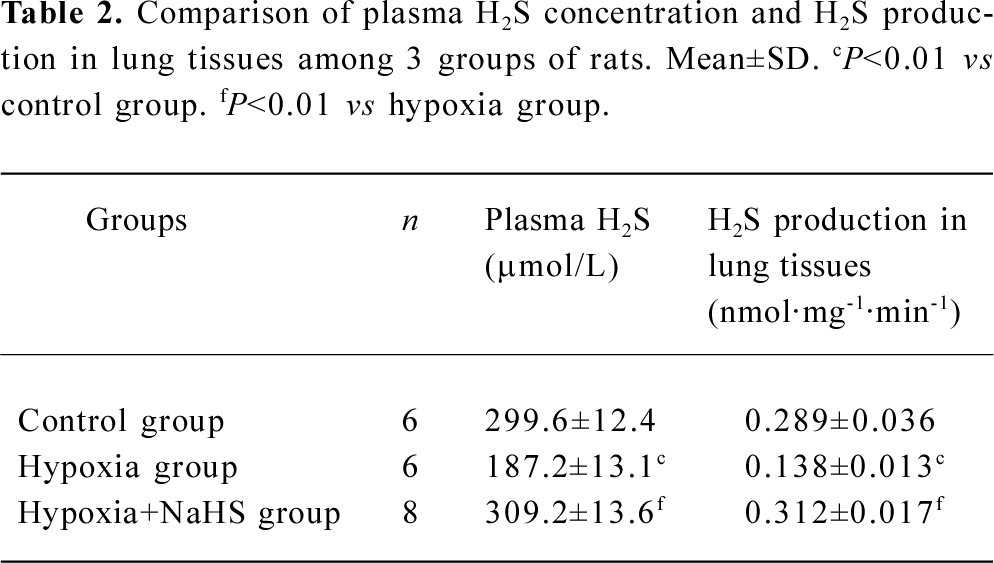
Full table
Oxidative stress indicators In the present study, we chose several oxidative injurants and antioxidants to evaluate the redox state in vivo. Our data showed that the GSSG of lungs in the rats of the hypoxia group was higher than those of the control group (0.026±0.005 vs 0.016±0.003 g/g, P<0.01). After we treated the hypoxia group with the donor of H2S, the GSSG level in the hypoxia+NaHS group was decreased by 23% compared with the hypoxia group (P<0.05). The MDA level increased obviously in the hypoxia group as compared with the control group (0.0054±0.0005 vs 0.0032±0.007 mmol/g, P<0.01). However, treatment with NaHS could not reverse the elevation of MDA under hypoxic challenge. The T-AOC of rat lung tissues in the hypoxia group was significantly lower than that of the control group (15361.2±221.6 vs 19578.6±2994.7 U/g, P<0.01). After administration with NaHS, however, the T-AOC was enhanced by 19% as compared with of the hypoxia group (P<0.05). For the other 2 antioxidants, our data showed that there were no significant differences among the 3 groups in lung SOD and GSH levels, as shown in Table 3.
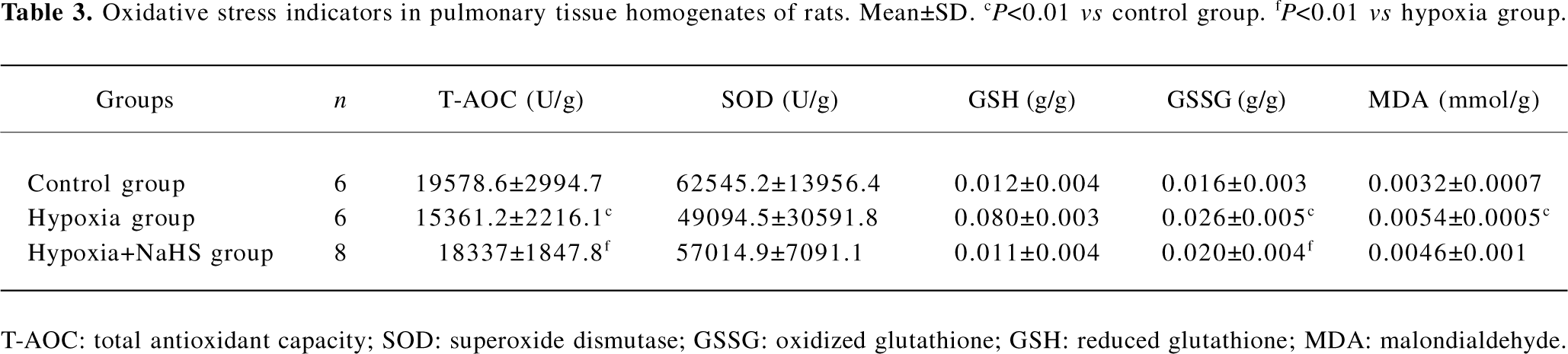
Full table
SOD mRNA level To explore the mechanism responsible for the possible role of H2S in the regulation of antioxidative response, we chose the most important antioxidant, SOD, in our further study. There were no significant differences, however, in the lung SOD1 and SOD2 mRNA levels among the rats of the 3 groups, as shown in Table 4.
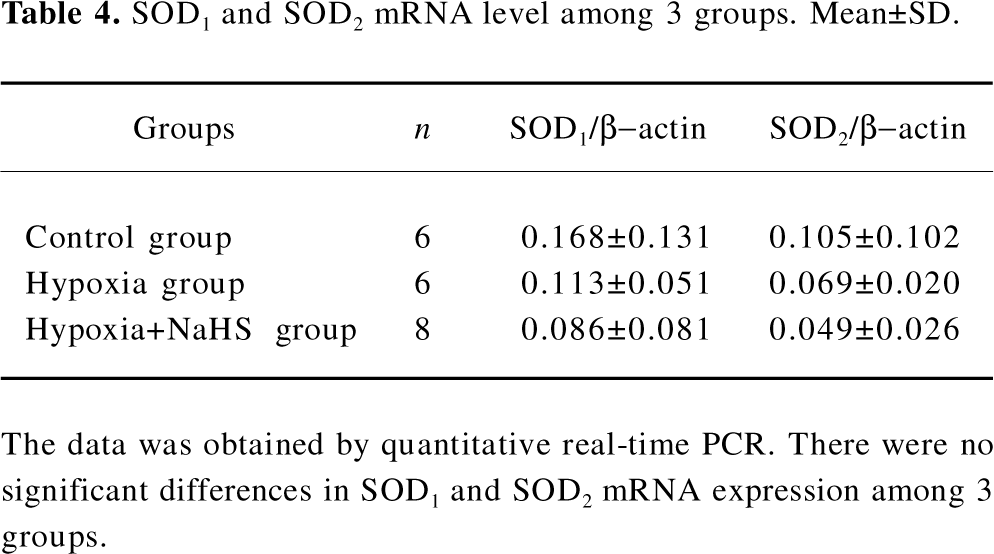
Full table
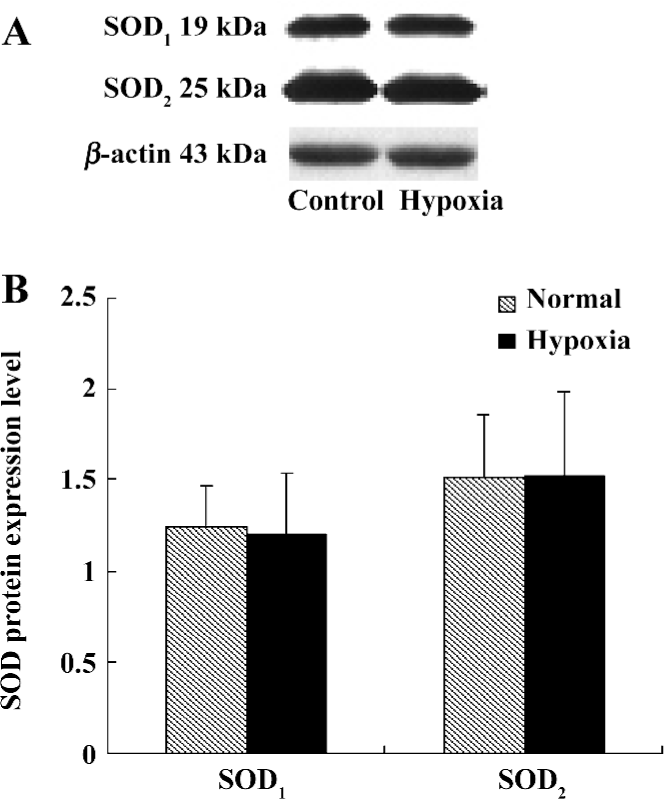
SOD protein level We also examined the SOD1 and SOD2 protein expression levels in the lung tissues of rats in both the control group and hypoxia group. The results showed that there were no distinct differences between these 2 groups, as seen in Table 5 and Figure 3.
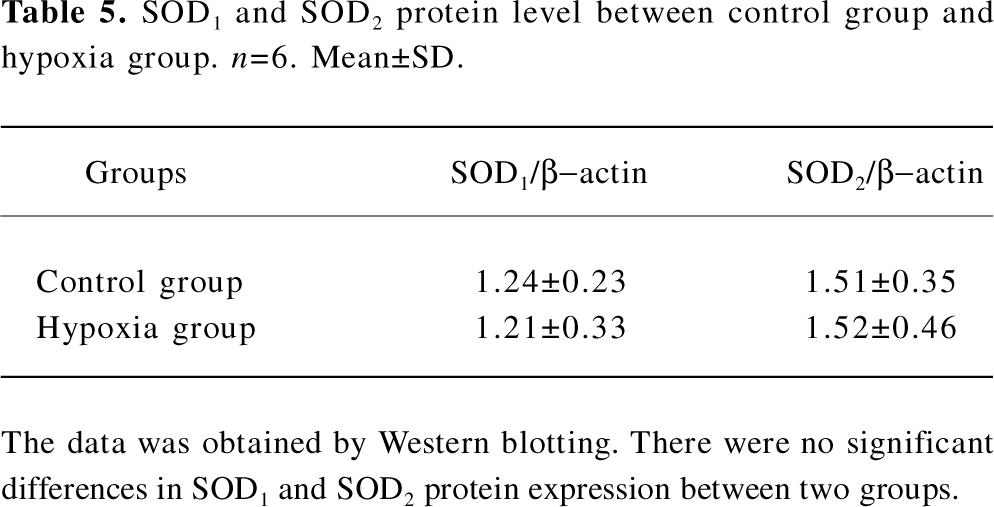
Full table
Discussion
Pulmonary hypertension is an important pathophysiological process of many hypoxic diseases, such as cardiopulmonary disease, mountain sickness, and sleep apnea syndrome[19]. However, the pathogenesis of hypoxic pulmonary hypertension has not been fully understood[20]. The overproduction of ROS can result in the progression of pulmonary vascular remodeling[15]. Recently, endogenous H2S has been suggested as a novel neuromodulator and signal transmitter[21]. Our previous work revealed that the endogenous CSE/H2S pathway was down-regulated in hypoxic pulmonary hypertension[14]. H2S could exert antioxidant effects in vivo and in vitro[17,22]. Based on these observations, we hypothesized that H2S might act as an antioxidant in HPH and protect the body from oxidative injury. The present study aimed to investigate the role of H2S in the pathogenesis of oxidative stress in HPH.
Pulmonary artery hypertension was defined as a mPAP of 25 mmHg or more and pulmonary capillary wedge pressure of 15 mmHg or less, both measured at rest by right heart catheterization according to WHO criteria[23]. Our previous work demonstrated that after intermittent exposure to 10% oxygen for 6 h daily for 2−4 weeks, the rats could develop constant chronic pulmonary hypertension with right ventricular hypertrophy[8,11,14,24]. In the present study, after 3 weeks of hypoxia, the mPAP increased significantly compared to the rats without hypoxia (P<0.01). The ratio of RV/(LV+SP) in the hypoxia group was also higher than that in the control group (P<0.01). These results showed that the rat model of HPH was reproduced successfully.
Long-term hypoxia can result in an increase in the rat plasma glutathione disulfide level in vivo and an increase in the formation of lipid peroxide products in cultured bovine pulmonary endothelial cells in vitro[15,25]. The overproduction of ROS can induce the progression of pulmonary vascular remodeling. Thus several intrinsic substances to modulate pulmonary ROS levels may play protecting or aggravating roles in the progression of hypoxic pulmonary vascular remodeling. As we know, oxidative stress is an imbalance state of the oxidative and antioxidative systems[26]. When the balance is disturbed, ROS, by promoting lipid peroxidation, may cause damage to DNA, proteins, lipid, and membranes in the cells[27]. In the present study, MDA level of lung tissues in the hypoxia group increased after hypoxia exposure for 3 weeks (Table 3). Antioxidants are the primary defense against ROS. In the present study, we choose to detect several antioxidants in lung tissues, such as T-AOC and SOD. To further investigate the cellular redox status, we used GSH/GSSG as the intracellular redox buffers. Our data showed that T-AOC in the hypoxia group was significantly lower than that of the control group (P<0.01), and GSSG in the hypoxia group was higher than that of the control group (P<0.01). There were no significant differences in SOD and GSH between the hypoxia group and control group. It is worth noting that in our study, when the pulmonary hypertension formed, MDA and GSSG increased, whereas T-AOC which was representative of total antioxidant capacity, quenched obviously. Based on these results, we speculated that oxidative stress occurred and lasted in the development of HPH, which might facilitate the progression of pulmonary vascular remodeling.
H2S is a strong reducing agent. It is a highly-reactive molecule and may easily react with other compounds, especially with ROS and nitrogen species[16,17]. Kimura Y and Kimura H found that H2S increased the glutathione levels, which normally decrease during the cell death cascade, by enhancing the activity of γ-glutamylcysteine synthetase and up-regulating cysteine transport. Their observation revealed that H2S protected neurons from oxytosis by increasing the production of antioxidant glutathione[15]. The down-regulation of the endogenous H2S pathway was involved in the pathogenesis of HPH in rats, and it was found that exogenously applied H2S could exert protective effects on the pathogenesis of HPH[14]. In present study, hypoxia-induced oxidative stress injury, accompanied by the H2S plasma level and its production in lung tissues, were all quenched significantly. To explore the effects of H2S on the regulation of oxidative stress, we administered the exogenous donor of H2S by injecting NaHS intraperitoneally. We observed that after the treatment with exogenous H2S, hypoxic pulmonary hypertension was attenuated, the T-AOC levels was increased by 19%, and GSSG content was decreased by 23%. These results suggested that H2S might play an important role in regulating oxidative stress in hypoxia-induced pulmonary hypertension, at least by directly scavenging GSSG and increasing the T-AOC. In the present study we first characterized the antioxidative aspect of H2S in the process of hypoxic pulmonary hypertension. An expanding body of experimental observations supports the concept that H2S might scavenge excess oxidative stress production[15,17,27]. However, the exact mechanism needs to be further investigated.
The SOD family is the key antioxidant enzyme during oxidative stress. Up to now, 3 SOD isoenzymes have been identified in mammals[28]. The major intracellular SOD is a copper- and zinc-containing homodimer (Cu/Zn SOD or SOD1) present throughout the cytoplasm and nucleus. The mitochondrial SOD (MnSOD or SOD2) is synthesized in the cytoplasm and translocated to the inner matrix of mitochondria. The last SOD discovered is primarily extracellular (EC-SOD or SOD3)[27]. In order to detect whether SOD changes during the oxidative stress of HPH, we investigated the activity and transcriptional level of SOD1 and SOD2. Our study showed that there was no significant difference in SOD1 and SOD2 among the 3 groups. However, the SOD3 level was not detected in our study. Whether extracellular SOD can exert important functions in HPH still remains to be answered.
In conclusion, oxidative stress was involved in hypoxic pulmonary hypertension. H2S could enhance the T-AOC and attenuate GSSG in the lung tissues of rats with HPH. The present study also showed that H2S had no effect on SOD mRNA or the protein level in the lung tissues of rats with chronic hypoxic pulmonary hypertension. These results suggest that H2S not only ameliorates the pathological changes of HPH, but also acts as a cardiovascular protective antioxidant in the development of HPH.
Author contribution
Hong-ling WEI, Chun-yu ZHANG, Hong-fang JIN, Chao-shu TANG designed research; Hong-ling WEI and Chun-yu ZHANG performed research; Jun-bao DU and Hong-fang JIN contributed new reagents or analytic tools; Hong-ling WEI, Jun-bao DU, Chun-yu ZHANG analyzed data; Hong-ling WEI and Jun-bao DU wrote the paper.
References
- Rhodes J. Comparative physiology of hypoxic pulmonary hypertension: historical clues from brisket disease. J Appl Physiol 2005;98:1092-100.
- Hoshikawa Y, Ono S, Suzuki S, Tanita T, Chida M, Song C, et al. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J Appl Physiol 2001;90:1299-306.
- Chandel NS, Maltepe E, Godlwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 1998; 95: 11 715–20.
- Waypa GB, Marks JD, Mack MM, Boriboun C, Mungai PT, Schumacker PT. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ Res 2002;91:7191-26.
- Waypa GB, Chandel NS, Schumacker PT. Model of hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res 2001;88:1259-66.
- Hiromitsu M, Tatsuo S, Kanami I, Xing GQ, Katsuyuki A, Toshiro F. Adrenomedullin can protect against pulmonary vascular remodeling induced by hypoxia. Circulation 2004;109:2246-51.
- Geng B, Yan H, Zhong GZ, Zhang CY, Chen XB, Jiang HF, et al. Hydrogen sulfide: a novel cardiovascular functional regulatory gas factor. Beijing Da Xue Xue Bao 2004;35:556-70.
- Zhang QY, Du JB, Zhou WJ, Yan H, Tang CS, Zhang CY. Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem Biophys Res Commun 2004;317:30-7.
- Ali MY, Ping CY, Mok YY, Ling L, Whiteman M, Bhatia M, et al. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulfide. Br J Pharmacol 2006;149:625-34.
- Jin HF, Du JB, Li XH, Wang YF, Liang YF, Tang CS. Interaction between hydrogen sulfide/cystathionine γ-lyase and carbon monoxide/heme oxygenase pathways in aortic smooth muscle cells. Acta Pharmacol Sin 2006;27:1561-6.
- Wang YF, Shi F, Du JB, Tang CS. Impact of L-arginine on hydrogen sulfide/cystathionine γ-lyase pathway in rats with high blood flow-induced pulmonary hypertension. Biochem Biophys Res Commun 2006;345:851-7.
- Yan H, Du JB, Tang CS. Possible effects of endogenous hydrogen sulfide on aortic remodeling in hypertensive rats. Beijing Da Xue Xue Bao 2004;36:234-7.
- Li XH, Du JB, Shi L, Li J, Tang XY, Qi JG, et al. Down-regulation of endogenous hydrogen sulfide pathway in pulmonary hypertension and pulmonary vascular structural remodeling induced by high pulmonary blood flow in rats. Circ J 2005;69:1418-24.
- Zhang CY, Du JB, Bu DF, Yan H, Tang XY, Tang CS. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem Biophys Res Commun 2003;302:810-6.
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J 2004;18:1165-76.
- Whiteman M, Cheung NS, Zhu YZ, Chu SH, Siau JL, Wong BS, et al. Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain. Biochem Biophys Res Commun 2005;326:794-8.
- Geng B, Chang L, Pan CS, Qi YF, Zhao J, Pang YZ, et al. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochem Biophys Res Commun 2004;318:756-63.
- Stipanuk MH, Beck PW. Characterization of the enzymatic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J 1982;206:267-77.
- Moudgil R, Michelakis ED, Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol 2005;98:390-403.
- Das M, Dempsey EC, Bouchey D, Reyland ME, Stenmark KR. Chronic hypoxia induces exaggerated growth responses in pulmonary artery advential fibroblasts. Am J Respir Cell Mol 2000;22:15-25.
- Lowicka E, Bettowski J. Hydrogen sulfide (H2S)––the third gas of interest for pharmacologists. Pharmacol Rep 2007;59:4-24.
- Yan SK, Chang TJ, Wang H, Wu LG, Wang R, Meng QH. Effects of hydrogen sulfide on homocysteine-induced oxidative stress in vascular smooth cells. Biochem Biophys Res Commun 2006;351:485-91.
- McGoon M, Gutterman D, Steen V, Barst R, McCrory DC, Fortin TA, et al. Effects of hydrogen sulfide on hypoxia pulmonary vascular structural remodeling. Chest 2004;126:S14-34.
- Jin HF, Cong BL, Zhao B, Zhang CY, Liu XM, Zhou WJ, et al. Effects of hydrogen sulfide on hypoxia pulmonary vascular structural remodeling. Life Sci 2006;78:1299-309.
- Janssen-Heininger MW, Persinger RL, Korn SH, Pantano C, Mcelhinney B, Reynaert NL, et al. Reactive nitrogen species and cell sigaling. Am J Respir Crit Care Med 2002;166:S9-16.
- Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol 2005;25:29-38.
- Liu HJ, Colavitti R, Rovira II, Finkel T. Redox-dependent transcriptional regulation. Circ Res 2005;97:967-74.
- Bowler RP, Crapo JD. Oxidative stress in airways: Is there a role for extracellular superoxide dismutase. Am J Respir Crit Care Med 2002;166:38-43.