
From dissection of disease pathogenesis to elucidation of mechanisms of targeted therapies: leukemia research in the genomic era
Introduction
Leukemia is one of the most notorious enemies of mankind and accounts for some 300 000 new cases and 222 000 deaths each year worldwide. It is the leading cause of cancer death among men under 40 years and among women under 20 years[1]. This high ratio of deaths/cases (74%) reflects the unsatisfactory prognosis of leukemia with current therapeutics, mainly composed of chemotherapy. Therefore, the development of new therapeutic approaches for leukemia patients is still an urgent need for scientists/hematologists, while dissection of leukemia pathogenesis is critical to the development of molecularly targeted therapies that might have maximal therapeutic efficacies with minimal adverse effects.
Recognition of leukemia: from morphological change to molecular markers
Leukemia represents a group of hematological malignancies characterized by clonal expansion of hematopoietic cells with uncontrolled proliferation, decreased apoptosis and blocked differentiation. According to the disease progression and hematopoietic lineages involved, leukemia can be divided into acute or chronic, lymphoid or myeloid, with a number of additional subtypes based on the distinct stages of differentiation block along with each lineage[2]. Acute myelogenous leukemia (AML), the most common leukemia diagnosed in adults, is a malignant disease of the bone marrow in which hematopoietic precursors are arrested in an early stage of myeloid development. AML leukemic cells can be identified in most instances by the presence of Auer rods, myeloperoxidase or monocyte-associated esterases, and AML can be divided into M0–M7 subtypes. Most subtypes are distinguished from other related blood disorders by the presence of more than 20%–30% blasts in the bone marrow. In contrast, acute lymphoid leukemia (ALL) is the most common cancer in childhood and leukemic lymphoblasts lack specific morphological or cytochemical features.
After three decades of rapid advances, leukemia research has generated a rich and complex body of knowledge, revealing leukemia to be a disease of hematopoietic stem/progenitor cells that involves dynamic changes in the genome. Recurrent chromosomal changes occur in more than half of all cases of leukemia, and more than 300 chromosomal translocations have been detected so far. The pathogenesis of AML involves an array of molecular alterations that disrupt almost every facet of cell transformation. These processes include the regulation of cell proliferation, differentiation, self-renewal, survival, cell cycle checkpoint control, DNA repair and chromatin stability, and cell dissemination[3]. A vastly improved understanding of ALL pathophysiology has emerged from two decades of progress in defining the lineage-related development, antigen expression and genetic abnormalities of leukemic cells, and in elucidating the multistep mechanisms by which changes in the function of specific genes disrupt key signaling pathways, which ultimately lead to leukemic transformation[4]. Tremendous advances in the elucidation of cytogenetic and molecular genetic abnormalities of major AML and ALL subtypes (Table 1) have not only enabled the classification of leukemia into progno-stically and therapeutically important distinct clinical entities, but have also identified candidate molecules for targeted therapeutics of AML and ALL.
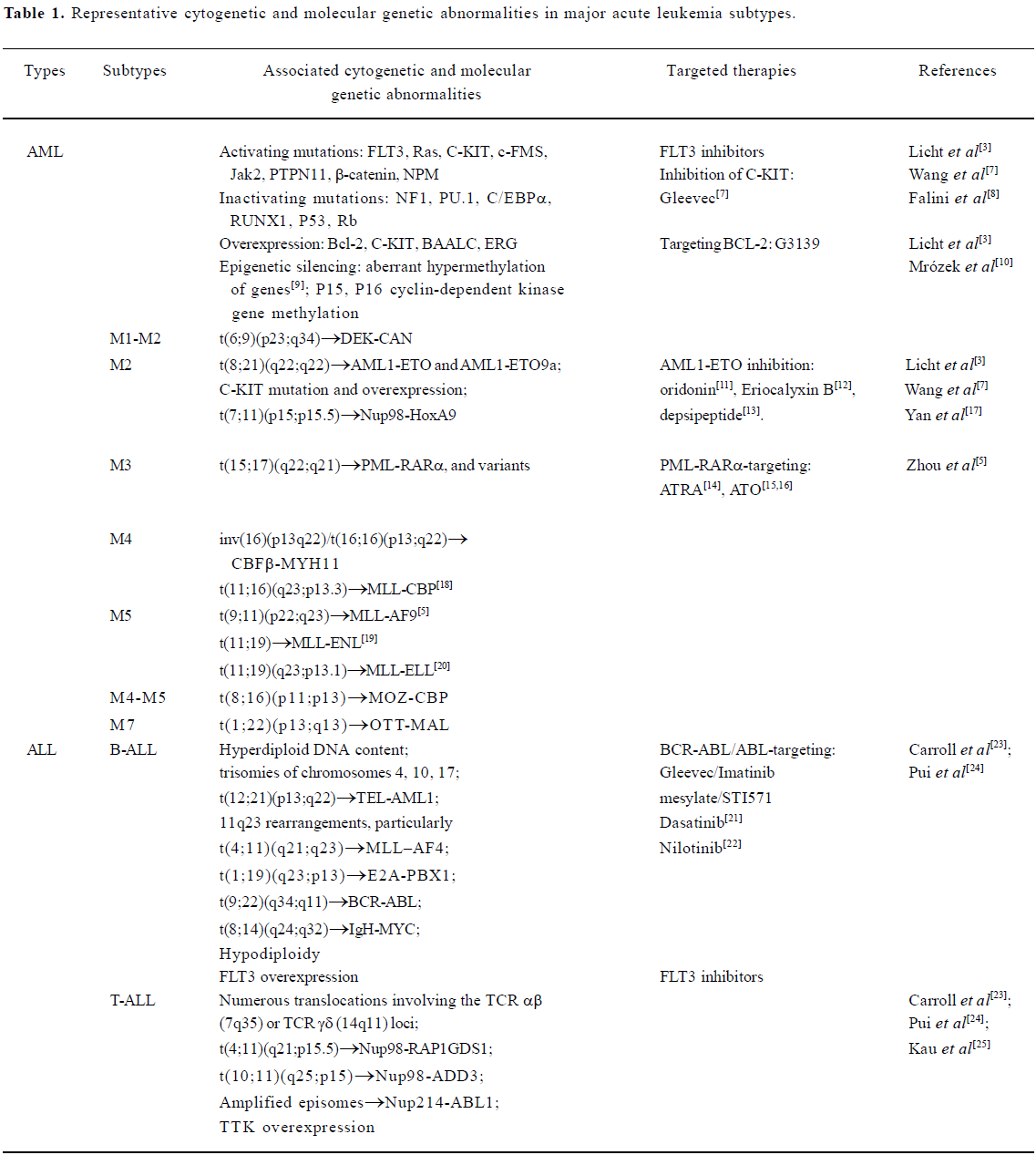
Full table
AML: recent advances
AML is a very heterogeneous disease with regard to clinical features and acquired genetic alterations, both those detected microscopically as structural and numerical chromosome aberrations, and those detected as submicroscopic gene mutations and changes in gene expression. In addition, the responses of AML to therapies differ from one subtype to another. Thus, to improve clinical outcome, the therapeutic strategies should be disease pathogenesis-based and individualized.
APL: from disease pathogenesis to targeted therapies
APL is characterized by the presence of an accumulation of abnormal promyelocytes in bone marrow or peripheral blood that do not differentiate into mature granulocytes, the occurrence of fibrinogenopenia and disseminated intravascular coagulation that is often worsened by chemotherapy, and the presence of the specific chromosomal translocation t(15;17)(q22;q21) or its variants[5].
t(15;17) and PML-RARα fusion gene Molecular cloning and sequence analysis of the genes involved in t(15;17) are critical for our understanding of the pathogenesis of APL and the differentiation and apoptosis therapies for this subtype of AML. Chen et al[6] reported that rearrangements in the second intron of the RARα gene on chromosome 17 are present in a large majority of APL patients and the 5’ part of the RARα gene has been shown to be frequently disrupted in Chinese patients with APL[26]. On chromosome 15, rearrangements of the MYL (named PML later) gene and a breakpoint cluster region as well as some molecular variants have been reported[27]. The entire PML genomic gene has been cloned and its exon-intron structure has also been established[28,29].
It is well established that the chromosomal translocation t(15;17) disrupts the PML gene on chromosome 15 and the RARα gene on chromosome 17, resulting in chimeric PML-RARα and RARα-PML fusion genes[30,31]. To characterize the expression patterns of the PML-RARα fusion gene, a sensitive reverse transcriptase-polymerase chain reaction (RT-PCR) procedure was established and was used to analyze the PML-RARα chimeric mRNAs in patients with APL[32,33]. Three distinct types of PML-RARα transcripts, long (L), short (S) and variant (V), were identified. Recently, a real-time RT-PCR system was established to quantify the dose of PML-RARα fusion transcripts in APL patients at distinct disease stages and this system was useful in reflecting leukemic burden, assessing response to treatment and indicating the ultimate clinical outcome or curability of disease[34].
To gain further insights into the molecular basis of t(15;17), Gu et al[35] sequenced the entire genomic DNA region of RARα. All previously reported “spacer” sequences in V-type transcripts[36] were found in intron 2 of the RARα gene and most of these sequences were flanked by gt splice donor sites. Two cases with a relatively long spacer sequ-ence showed resistance to RA treatment. In these cases, the aberrant V-type PML-RARα protein displayed increased affinity to the nuclear corepressor protein SMRT, providing further evidence that RA exerts a therapeutic effect on APL through modulation of the RARα–corepressor interaction. Among patients with the L-type or S-type PML-RARα fusion transcript, some consensus motifs were identified at the hotspots of the chromosome 17q breakpoints within intron 2 of –RARα, strengthening the importance of this intron in the pathogenesis of APL.
t(11;17)(q23;q21) and PLZF-RARα fusion gene A subset of APL cases were shown to present atypical phenotypic, cytogenetic or molecular features at different stages of the disease[37]. In 1993, Chen et al[38] reported an unusual karyotype 46, XY, t(11;17)(q23;q21) and showed a rearrangement between the RARα gene and a newly discovered promyelocytic leukemia zinc finger (PLZF) gene. Chen et al[39] demonstrated that the PLZF gene encodes a potential transcription factor containing nine zinc finger motifs related to the Drosophila gap gene Kruppel and is expressed as at least two isoforms that differ in the sequences encoding the N-terminal region of the protein. The PLZF gene was 201-kb long and contained six exons and five introns. At least four alternative splicings (AS-I, -II, -III, and -IV) were found within exon 1. These alternative splicings maintained the open reading frames (ORF) and may encode isoforms with an absence of important functional domains. A TATA box and several transcription factor binding sites were found in the putative promoter region upstream of the transcription start site[40]. Like the PML-RARα protein, PLZF-RARα inhibits ligand-dependent transactivation of RARα, reflecting the “dominant negative” effect of PLZF-RARα on wild-type RARα and implicating the PLZF-RARα fusion protein in the molecular pathogenesis of APL[41].
Variant chromosomal translocations were also identified in APL, that is, t(5;17)(q35;q21) generating NPM-RARα, t(11;17)(q13;q21) resulting in NuMA-RARα and dup(17)(q11;q21) with generation of Stat5b-RARα fusions[42].
APL murine models Transgenic mice were generated to study the leukemogenic potential of the fusion genes in vivo. Mice expressing the human cathepsin G (hCG)-PML-RARα transgene were found to have altered myeloid development that was characterized by increased percentages of immature and mature myeloid cells in the peripheral blood, bone marrow and spleen. In addition, approximately 30% of transgene-expressing mice eventually developed acute myeloid leukemia after a long latent period[43]. Cheng et al[44] generated transgenic mice with PLZF-RARα and NPM-RARα. PLZF-RARα transgenic animals developed chronic myeloid leukemia (CML)-like phenotypes, whereas NPM-RARα transgenic mice showed a spectrum of phenotypes from typical APL to CML. In contrast to bone marrow cells from PLZF-RARα transgenic mice, those from NPM-RARα transgenic mice could be induced to differentiate by all-trans retinoic acid (ATRA). Dissociation of SMRT from different receptors was also observed at different concentrations of ATRA for RARα-RXRα, NPM-RARα and PML-RARα, but was not observed in PLZF-RARα. These data clearly establish the leukemogenic role of PLZF-RARα and NPM-RARα and the importance of fusion receptor/corepressor interactions in the pathogenesis as well as in determining different clinical phenotypes of APL. Recently, RARα-PLZF and PLZF-RARα/RARα-PLZF double transgenic mice have also been generated[45].
Mechanisms of retinoic acid and arsenic treatment APL is the first example of a human cancer that can be effectively treated with the differentiation inducer ATRA, a derivative of vitamin A[14]. Nevertheless, arsenic trioxide (ATO) has been shown in China to have a strong therapeutic effect against APL through induction of apoptosis at high concentra- tion and partial differentiation at low concentration[15,16,46–49]. Modulation and degradation of PML-RARα fusion protein can be induced by both ATRA and ATO, demonstrating that APL is the first leukemia in which targeted therapy has proven to be effective[5,42,50–56]. Under a pharmacological con-centration (10-7–10-6 mol/L) of ATRA, the corepressors and histone deacetylase (HDAC) were released from the PML-RARα homodimer, while co-activator and histone acetylase were recruited, resulting in the relief of transcriptional repression. It has been shown that although the interaction with corepressors and HDAC of the RARα moiety in PLZF-RARα was modulable under 10-6 mol/L ATRA, binding to the corepressor complex on the N-terminal POZ domain of the PLZF moiety was retained even under a very high ligand concentration (10-5 mol/L). As a result, ATRA alone cannot induce maturation of PLZF-RARα-harboring cells, whereas HDAC inhibitor is required to cooperate with ATRA to induce differentiation of these cells[36]. More recently, proteolysis of PML-RARα via different pathways has drawn considerable attention. Although it was reported that ATRA could trigger caspase-mediated cleavage of the PML-RARα chimeric protein on PML[57], further dissecting of the pathways involved in PML-RARα catabolism led to the discovery of ubiquitin/proteasome system (UPS)-mediated degradation of PML-RARα and RARα, which was dependent on the binding of SUG-1 in the AF2 transactivation domain of RARα with 19S proteasome[58,59]. The degradation of PML-RARα contributes to the restoration of normal retinoid signaling and PML-NB functions, although this event seems to be relatively late compared to the modulation of chimeric protein transregulatory activity. ATRA also induces cAMP, a differentiation enhancer that boosts transcriptional activa-tion, reverses the silencing of the transactivating function of RXR, restores ATRA-triggered differentiation in mutant ATRA-resistant cells[60] and inhibits cell growth by modulating several major players in G(1)/S transition regulation. Garzon et al[61] demonstrated that ATRA treatment of APL patients and cell lines modulates a small number of micro-RNAs (miRNA), most of which have confirmed targets involved in hematopoietic differentiation and apoptosis. They found that along with miR-223, miR-107 negatively regulated NFI-A; ATRA induced NF-κB bond to the promoter of let-7a-3 and activated its transcription; and ATRA down-regulation of RAS and Bcl-2 correlated with the activation of two confirmed miRNA regulators of these genes, let-7a- and miR-15a/miR-16-1, respectively. Systems analysis of transcriptome and proteome in ATRA-induced APL cell differentiation reveals induction of an array of transcription factors and cofactors, activation of calcium signaling, stimulation of the IFN pathway, activation of the UPS necessary for degradation of the PML-RARα and restoration of the PML-NB, cell cycle arrest, induction of cyclooxygenase 1[62], inhibition of angiogenesis[63], down-regulation of tissue factor[64], and a gain in apoptotic potential. Of note, a number of novel genes, RA-induced genes (RIG, such as RIG-G, E, K, and I), have been cloned at the Shanghai Institute of Hematology and have been shown to have very interesting functional features[65–69].
The cysteine-rich region of the PML protein sequence is a principal candidate for interaction with trivalent arsenic. ATO induces modulation and catabolism of the PML-RARα fusion protein in a pattern that differs from that induced by ATRA. ATO treatment causes APL cells to experience a series of changes, including reaggregation of PML-NB antigens, recruitment of PML-RARα proteins onto NB and degradation of PML-RARα[70]. That ATO targets the PML moiety of PML-RARα is supported by the observation that a similar modulation process of wild-type PML, but not RARα, occurs in APL or non-APL cells. Through a yet unknown mechanism, ATO causes PML to be located to the nuclear matrix and sumoylated at two important residues resulting in different consequences, that is, sumolyation at K160 necessary for 11S proteasome recruitment and subsequent sumolyation at K490 for nuclear localization and degradation[71,72]. ATO treatment also induced phosphorylation of the PML protein through a mitogen-activated protein (MAP) kinase pathway[73].
A striking similarity in the effect of the two otherwise unrelated agents, ATRA and ATO, is the degradation of PML-RARα oncoprotein through distinct pathways[5,42,74]. Intriguingly, a clearance of PML–RARα transcript in an earlier and more thorough manner, and higher-quality remission and survival in newly diagnosed APL are achieved when ATRA is combined with ATO compared with either monotherapy, making APL a curable disease[75,76]. To gain insights into the synergic effects of the ATRA/ATO combination on APL cells, systems analysis of transcriptome and proteome has been carried out in RA/ATO-induced cell differentiation/apoptosis. Zheng et al[77] reported that at an early state (within 6 h), ATRA plus ATO modulated transcription factors/cofactors associated with myeloid-specific gene expression, nuclear receptor signaling molecules, interferon pathway members and factors involved in some other cascades. At a time point of 12–24 h, ATRA/ATO-regulated genes/proteins seemed to be an amplification of RA signaling and a strong activation of the ubiquitin/proteasome system, which might facilitate degradation of PML-RARα. After 48–72 h of treatment with RA/ATO, the expression of the differentiation markers and functional molecules reached a maximum, whereas genes/proteins promoting cell cycle or enhancing cell proliferation were significantly repressed. As the cells approached terminal differen-tiation, the expression of apoptosis agonists increased gradually. Recently Leung et al[78] reported that treatment with ATRA led to a dose-dependent increase in aquagly-ceroporin 9 (AQP9) gene transcription and protein expres-sion, which in turn increased arsenic uptake into the cells. These processes might contribute to the mechanisms of ATRA/ATO-induced differentiation/apoptosis of APL cells.
AML M2 with t(8;21)
AML M2, or myeloblastic leukemia with maturation, is one of the most common types of AML and occurs in about 25% of all cases. A characteristic translocation observed in AML M2 is t(8;21). Treatment includes intensive multidrug chemotherapy and in selected cases allogeneic bone marrow transplantation. Nevertheless, the outcome for AML M2 patients remains poor with an overall survival of 35%–60%[79]. Children with AML M2 carrying the t(8;21) translocation have a better prognosis (69% survival). New therapeutics are required to increase the probability of cure in this serious disorder.
Translocation t(8;21) and resultant AML1-ETO fusion The t(8;21)(q22;q22), where coding sequences of the AML1 gene on chromosome 21 are juxtaposed to coding sequences of the ETO gene on chromosome 8, generating an AML1-ETO (AE) fusion transcript, is seen in 40%–80% of AML M2 and 12%–20% of all cases of AML[80,81], and represents the most common chromosomal translocation in AML[80,82]. The AE fusion protein recruits the N-CoR-mSin3–HDAC complex[83,84], inhibits transcription of AML1 target genes[80,83], including interleukin-3 (IL-3)[85] and granulocyte-macrophage colony-stimulating factor (GM-CSF)[86,87], activates transcription of apoptotic antagonist Bcl-2[88], upregulates protein tyrosine kinase C-KIT[7], induces the expression of granulocyte colony-stimulating factor receptor (G-CSFR) as well as myeloperoxidase (MPO)[89], and blocks transactivation of the GM-CSF promoter[87]. The AE oncoprotein enhances self-renewal of hematopoietic stem/progenitor cells, blocks hematopoietic differentiation, disturbs normal cell proliferation[82] and immortalizes murine hematopoietic progenitors[90,91]. Although reports suggest that additional mutations are required to cooperate with AE to cause murine full-blown leukemia[90,92], stem cells expressing AE induce a myeloproliferative disorder[93] or distinct myeloid developmental abnormalities in mice[94], while deletion of a C-terminal NcoR/SMRT-interacting region strongly induces leukemia development[95]. Recently, a novel isoform of the AE transcript, AML1-ETO9a (AE9a), which includes an extra exon 9a of the ETO gene and encodes a C-terminally truncated AE protein, was identified from human t(8;21) AML and was shown to rapidly induce leukemia in a mouse retroviral transduction–transplantation model[17]. These data demonstrate that AE plays a critical role in the pathogenesis of t(8;21) leukemia and that AE-targeting agents might be helpful for the treatment of t(8;21) AML.
Abnormalities of receptor tyrosine kinases in t(8;21) AML To further explore the genetic abnormalities in AML M2 with t(8;21), Wang et al[7] screened a number of candidate genes and identified 11 types of mutations in the C-KIT gene (mC-KIT), including 6 previously undescribed ones, among 26 of 54 (48.1%) cases with t(8;21). To address a possible chronological order between AE and mC-KIT, we showed that among patients with AE and mC-KIT most leukemic cells at disease presentation harbored both genetic alterations, whereas in three cases investigated during complete remission, only AE, but not mC-KIT, could be detected using allele-specific PCR. Therefore, mC-KIT should be a subsequent event on the basis of t(8;21). Furthermore, induced expression of AE in U937-A/E cells significantly upregulated mRNA and protein levels of C-KIT. This may lead to an alternative way of C-KIT activation and may explain the significantly higher C-KIT expression in 81.3% of patients with t(8;21) compared with patients with other leukemias. These data strongly suggest that t(8;21) AML follows a stepwise model in leukemo-genesis, that is, AE represents the first fundamental genetic hit to initiate the disease and activation of the C-KIT pathway may be the second, but also crucial, hit for the development of a full-blown leukemia.
Schessl et al[96] reported that AE occurred frequently together with activating mutations involving signal transduction pathways in patients with AML. To characterize genetic alterations that occur together with the AE fusion gene in AML, 135 patients with AE (93 men, 42 women; median age 50.9 years, range 15.8–89.1 years) were screened for activating mutations in the receptor tyrosine kinases FLT3 and C-KIT, as well as in NRAS (KITD816, NRAS codon 12/13/61). Patients included 118 with newly diagnosed AML, 4 in first relapse and 13 classified as having therapy-related AML. Activating mutations were detected in 38 patients (28.1%) and included mutations in the receptor tyrosine kinase FLT3 or C-KIT (25 patients in total) or in NRAS (13 patients). In contrast, no MLL-partial tandem duplication (MLL-PTD) mutations were detected in 87 samples subjected to this analysis. These data demonstrated that genetic alterations occurring with the AE fusion gene frequently affect signal transduction pathways.
Murine models for AML M2 The AE knock-in mice and transgenic mice using MRP promoter, a tetracycline-inducible system or Cre/LoxP system and lethally irradiated mice reconstituted by AE-expressing HSPC do not develop any malignancies[90,92,97,98] or leukemia[94,99], whereas Fenske et al[93] reported that targeted expression of the AE in mice using the Ly6A (Sca1) locus induces a spontaneous myeloproliferative disorder (MPD). With the presence of additional mutations, for example, mutagen N-ethyl-N- nitrosourea (ENU), introduced mutations or coexpression of TEL-PDGFRβ fusion gene, exogenous AE-expressing mice develop an AML phenotype[90,92,100]. Schessl et al[96] reported that AE collaborated with FLT3-length mutation to induce acute leukemia in a murine BM transplantation model. These data strongly suggest that additional genetic alterations are required to cooperate with full length AE in inducing leukemia.
To investigate the nature of the secondary mutations required for AE-mediated leukemogenesis, Yan et al[95] carried out bone marrow transplant experiments by using hematopoietic cells infected with an AE-expressing retrovirus. Interestingly, they found that in the absence of any artificially induced mutations, one mouse rapidly developed AML 14 weeks after the bone marrow transplantation[95]. A surprising finding that emerged from this study was that, because of a 1-bp insertion, the leukemic cells expressed a C-terminal 200-aa truncated form of AE (AEtr) lacking a critical domain for NCoR/SMRT and ETO interaction. Unlike its full-length counterpart, the expression of AEtr resulted in a rapid onset of leukemia in transplantation-recipient mice. The associated growth-arrest function of AE was lost with AEtr. In addition, the regulation of cell-cycle regulatory proteins, such as cyclin D3, cyclin A, CDK4, and CDK inhibitors p21WAF1/CIP1 and p27KIP1, by AE and C-terminal truncated AE were opposed. These results indicated that disruption of molecular events relating to the function of the AE C-terminal NCoR/SMRT interacting domain promoted the onset of t(8;21)-involved leukemogenesis and offered a paradigm to the study of genetic events related to cancer development. Recently, Yan et al[17] reported the identification of a previously unknown alternatively spliced isoform of the AE transcript, AML1-ETO9a (AE9a), that included an extra exon, exon 9a, of the ETO gene. AE9a encodes a C-terminally truncated AE protein of 575 amino acids. Expression of AE9a leads to rapid development of leukemia in a mouse retroviral transduction–transplantation model. More importantly, coexpression of AE and AE9a results in the substantially earlier onset of AML and blocks myeloid cell differentiation at a more immature stage. These results indicate that fusion proteins from alternatively spliced isoforms of a chromosomal translocation may work together to induce cancer development.
Potential targeted therapies for t(8;21) AML Clinically, aggressive cytosine arabinoside (Ara-C)-based chemotherapy is the standard protocol for t(8;21) AML, and t(8;21) has been shown to be a favorable prognostic factor for AML[101]. However, other researchers have demonstrated that the median survival time of patients with t(8;21) AML is less than 2 years with a 5-year survival rate of no more than 40%[7,102–106]. To further improve the clinical outcome and to provide therapeutic options for t(8;21) AML patients, investigational therapy should be developed. Zhou et al[11] reported that oridonin, a diterpenoid extracted from the medicinal herb Rabdosia rubescens, induced apoptosis of t(8;21) AML cells. Intriguingly, the t(8;21) product, AE fusion protein that plays a critical role in leukemogenesis, was degraded with generation of a catabolic fragment, whereas the expression pattern of the AE target genes investigated could be reprogrammed. The ectopic expression of AE enhanced the apoptotic effect of oridonin in U937 cells. Pre-incubation with caspase inhibitors blocked oridonin-triggered cleavage of AE, whereas substitution of Ala for Asp at residues 188 in the ETO moiety of the fusion abrogated AE degradation. Furthermore, oridonin prolonged the lifespan of C57 mice bearing truncated AE-expressing leukemic cells without suppression of bone marrow or reduction of body weight of animals, and exerted synergic effects when combined with cytosine arabinoside. Oridonin also inhibited tumor growth in nude mice inoculated with t(8;21)-harboring Kasumi-1 cells. These results suggest that oridonin may be a potential anti-leukemia agent that targets AE oncoprotein at residue D188 with low adverse effects, and may be helpful for the treatment of patients with t(8;21) AML. Furthermore, a genotype–phenotype correlation was revealed in terms of the response to tyrosine kinase inhibitor Gleevec/Imatinib meslate/STI571, in that the drug suppressed the C-KIT activity and induced proliferation inhibition and apoptosis in t(8;21) leukemic cells bearing the C-KIT N822K mutation or C-KIT overexpression, but not in cells with mC-KIT at residue D816. Gleevec also exerted a synergic effect on apoptosis induction with cytarabine, providing a potential use in the treatment of t(8;21) leukemia[7]. Yang et al[13] showed that AE undergoes degradation in response to treatment with histone deacety-lase inhibitors, one of which, depsipeptide (DEP), is currently undergoing Phase II clinical testing in a variety of malignancies. These compounds induce turnover of AE without affecting the stability of AE partner proteins. In addition, AE physically interacts with heat shock protein 90 (HSP90). DEP treatment interrupts the association of AE with HSP90 and induces proteasomal degradation of AE. DEP and the HSP90 antagonist 17-allylamino-geldanamycin (17-AAG) both triggered AE degradation, but without any additive or cooperative effects. These findings may stimulate the development of more rational and effective approaches for treating t(8;21) patients using histone deacetylase inhibitors or HSP90 inhibitors.
A mini-review on the advances in other sub-types of AML
Inv(16)(p13q22) is associated with AML M4Eo, which is characterized by the presence of myelomonocytic blasts and atypical eosinophils. This chromosomal rearrangement results in the fusion of CBFβ and MYH11 genes. CBFbeta normally interacts with RUNX1 (AML1) to form a transcriptionally active nuclear complex. Knockout of CBFbeta causes embryonic lethality because of a lack of definitive hematopoiesis. The MYH11 gene encodes the smooth muscle myosin heavy chain (SMMHC). The CBFbeta-SMMHC fusion protein is capable of binding to RUNX1 and forms dimers and multimers through its myosin tail. Results from transgenic mouse models show that CBFβ-MYH11 is able to inhibit predominantly RUNX1 function in hematopoiesis, and is a key player in the pathogenesis of leukemia[107]. The myeloid transcription factor CCAAT/enhancer-binding protein alpha (CEBPα) is crucial for normal granulopoiesis. Alterations to the structure and expression of CEBPα have been implicated in particular subtypes of AML. Helbling et al[108] found that conditional expression of CBFβ-SMMHC in U937 cells suppressed CEBPα protein expression and binding activity. However, CEBPα mRNA levels remained unchanged. No differences were detected in CEBPα mRNA levels in patients with inv(16) AML M4Eo (n=12) compared to patients with AML with a normal karyotype and M4 subtype (n=6), whereas CEBPα protein and binding activity were significantly reduced in patients with CBFβ-SMMHC. Furthermore, calreticulin, an inhibitor of CEBPα translation, was induced on mRNA and protein level in AML patients with CBFβ-SMMHC and after expression of CBFβ-SMMHC in the U937-cell system. Inhibition of calreticulin by siRNA restored CEBPα levels. These results suggest that modulation of CEBPα by calreticulin represents a novel mechanism involved in the differentiation block in CBFβ-SMMHC AML. Recently, Wunderlich et al[109] introduced the CBFβ-MYH11 cDNA into human CD34+ cells via retroviral transduction. Transduced cells displayed an initial repression of progenitor activity but eventually dominated the culture, resulting in the proliferation of clonal populations for up to 7 months. Long-term cultures displayed a myelomonocytic morphology while retaining multilineage progenitor activity and engraftment in NOD/SCID-B2M-/- mice. Progenitor cells from long-term cultures showed altered expression of genes defining inv(16) identified in microarray studies of human patient samples. This system will be useful in examining the effects of CBFbeta-SMMHC on gene expression in the human preleukemic cell, in characterizing the effect of this oncogene on human stem cell biology, and in defining its contribution to the development of leukemia.
The MLL-AF9 oncogene – one of the most frequent MLL/HRX/ALL-1 rearrangements found in infantile and therapy-related leukaemias – originates from t(9;11)(p22;q23) and is mainly associated with monocytic AML (AML M5). Pession et al[110] investigated the MLL-AF9 function using an antisense phosphorothioate-oligodeoxyribonucle-otide (MLL-AF9-PS-ODNas) and a THP-1 AML-M5 cell line carrying t(9;11). Having confirmed that MLL-AF9-PS-ODNas induced a strong inhibition of THP-1 cell growth and a moderate increase in apoptosis, they found that MLL-AF9-PS-ODNas did not induce morpho-functional terminal differentiation or restore M-CSF-, G-CSF- or GM-CSF-induced differentiation. Moreover, THP-1 cells showed the same phenotype with/without MLL-AF9-PS-ODNas. In THP-1 cells differentiated to mature macrophage-like cells by PMA/TPA or ATRA, MLL-AF9 expression was down-regulated. Thus, in the monocytic lineage, MLL-AF9 may be expressed only in early phases and can induce deregulated amplification in both non-malignant and malignant cells, maintaining the monocytic phenotype without blocking final maturation. Barabe et al[111] showed that after transplantation into immunodeficient mice, primitive human hematopoietic cells expressing MLL-AF9 or MLL-ENL generated myeloid or lymphoid acute leukemias, with features that recapitulated the human diseases. An analysis of serially transplanted mice revealed that the disease is sustained by leukemia-initiating cells (L-IC) that have evolved over time from a primitive cell type with a germline immunoglobulin heavy chain (IgH) gene configuration to a cell type containing rearranged IgH genes. The L-IC retained both myeloid and lymphoid lineage potential and remained responsive to microenvironmental cues. The properties of these cells provide a biological basis for several clinical hallmarks of MLL leukemias. By using a mouse model of human AML induced by the MLL-AF9 oncogene, Somervaille et al[112] demonstrated that colony-forming cells (CFC) in the bone marrow and spleen of leukemic mice were also leukemia stem cells (LSC). These self-renewing cells: (1) were frequent, accounting for 25%–30% of myeloid lineage cells at late-stage disease; (2) generated a phenotypic, morphological and functional leukemia cell hierarchy; (3) expressed mature myeloid lineage-specific antigens; and (4) exhibited altered microenvironmental interactions by comparison with the oncogene-immortalized CFC that initiated the disease. Krivtsov et al[113] isolated LSC from leukemias initiated in committed granulocyte macrophage progenitors through introduction of the MLL-AF9 fusion protein. The LSC were capable of transferring leukemia to secondary recipient mice when only 4 cells were transferred, and possessed an immunophenotype and global gene expression profile very similar to that of normal granulocyte macrophage progenitors. However, a subset of genes highly expressed in normal hematopoietic stem cells was re-activated in LSC. Thus, LSC can be generated from committed progenitors without widespread reprogramming of gene expression, and a leukemia self-renewal-associated signature is activated in the process. These findings define progression from normal progenitor to cancer stem cell and suggest that targeting a self-renewal program expressed in an abnormal context may be possible. The LSC responsible for sustaining, expanding and regenerating MLL-AF9 AML are downstream myeloid lineage cells, which have acquired an aberrant Hox-associated self-renewal program as well as other biological features of hematopoietic stem cells.
Interstitial loss of all or part of the long arm of chromosome 5, or del(5q), is a frequent clonal chromosomal abnormality in human myelodysplastic syndrome (MDS), a preleukemic disorder, and AML, and is thought to contribute to the pathogenesis of these diseases by deleting one or more tumor-suppressor genes. Although a major commonly deleted region (CDR) has been delineated on chromosome band 5q31.1, attempts to identify tumor suppressors within this band have been unsuccessful. Liu et al[114] focused their analysis of gene expression on RNA from primitive leukemia-initiating cells, which harbored 5q deletions, and analyzed 12 genes within the CDR that were expressed by normal hematopoietic stem cells. They found that the gene encoding alpha-catenin (CTNNA1) was expressed at a much lower level in leukemia-initiating stem cells from individuals with AML or MDS with a 5q deletion than in individuals with MDS or AML lacking a 5q deletion or in normal hematopoietic stem cells. Analysis of HL-60 cells, a myeloid leukemia line with deletion of the 5q31 region, showed that the CTNNA1 promoter of the retained allele was suppressed by both methylation and histone deacetylation. Restoration of CTNNA1 expression in HL-60 cells resulted in reduced proliferation and apoptotic cell death. Thus, loss of expression of the alpha-catenin tumor suppressor in hematopoietic stem cells may provide a growth advantage that contributes to human MDS or AML with del(5q).
ALL
Chromosomal aberrations are a hallmark of ALL, but alone fail to induce leukemia. To identify cooperating oncogenic lesions, Mullighan et al[115] carried out a genome-wide analysis of leukemic cells from 242 pediatric ALL patients using high-resolution, single-nucleotide polymorphism arrays and genomic DNA sequencing. Their analyses revealed deletion, amplification, point mutation and structural rearrangement in genes encoding principal regulators of B lymphocyte development and differentiation in 40% of B-progenitor ALL cases. The PAX5 gene was the most frequent target of somatic mutation, and was altered in 31.7% of cases. The identified PAX5 mutations resulted in reduced levels of PAX5 protein or the generation of hypomorphic alleles. Deletions were also detected in TCF3 (also known as E2A), EBF1, LEF1, IKZF1 (IKAROS) and IKZF3 (AIOLOS). These findings suggest that direct disruption of pathways controlling B-cell development and differentiation contributes to B-progenitor ALL pathogenesis. Moreover, these data demonstrate the power of high-resolution, genome-wide approaches in identifying new molecular lesions in cancer.
The NOTCH signaling pathway is essential in T-cell development and NOTCH1 mutations are frequently present in T-cell acute lymphoblastic leukemia (T-ALL). To gain insight into its clinical significance, NOTCH1 mutation was investigated in 77 patients with T-ALL[116]. Detection of NOTCH1 mutation was done using RT-PCR amplification and direct sequencing and compared with the clinical/biological data of the patients. Thirty-two mutations were identified in 29 patients (with dual mutations in 3 cases), involving not only the heterodimerization and proline/glutamic acid/serine/threonine domains as previously reported, but also the transcription activation and ankyrin repeat domains revealed for the first time. These mutations were significantly associated with elevated white blood cell counts at diagnosis and independently linked to short survival time. Interestingly, the statistically significant difference in survival according to NOTCH1 mutations was only observed in adult patients (>18 years) and not in pediatric patients (≤18 years), possibly because of the relatively good overall response of childhood T-ALL to the current chemotherapy. NOTCH1 mutations could coexist with HOX11, HOX11L2, or SIL-TAL1 expression. The negative effect of NOTCH1 mutation on prognosis was potentiated by HOX11L2 but was attenuated by HOX11. These data suggest that NOTCH1 mutation is an important prognostic marker in T-ALL and its predictive value could be even further increased if coevalu-ated with other T-cell-related regulatory genes. Thus, the NOTCH pathway acts in combination with oncogenic transcriptional factors on T-ALL pathogenesis.
Optimal use of antileukemic agents together with the stringent application of prognostic factors for risk-directed therapy in clinical trials has resulted in a steady improvement in the treatment outcome of ALL[24]. The future of treatment for ALL resides in defining the molecular pathways underlying the pathogenesis of this disease and in further elucidating the pharmacogenetic factors of the host. Improved understanding of the pathological features of the disease could in turn lead to the identification of new drugs or targets for specific treatments and may even suggest strategies for disease prevention. Selective potential targeted therapies for ALL are listed in Table 2.

Studies on chronic myelogenous leukemia
CML is a clonal myeloproliferative disorder characterized by genomic instability leading to its inevitable clinical evolution from an easily controlled chronic phase (CP) to a terminal blastic phase. The molecular mechanisms involved in blastic crisis remain largely unknown. Monoallelic deletions of the P53 gene in Chinese patients with CML in blastic crisis have been reported[118]. A few CP patients had additional abnormalities while most patients with disease progression [accelerated phase (AP) and blastic crisis (BC)] showed extra numerical and/or structural chromosomal aberrations[119,120].
STI571/Imatinib/Gleevec, a rationally designed agent that occupies the ATP-binding site of BCR-ABL and stabilizes the protein in its closed, inactive conformation, has become the new gold standard for treatment of CML[121–124]. However, its effects on patients with AP/BC are unsatisfactory because many patients relapse after transient remis-sion[125–127]. In addition, even among patients at CP, imatinib seems unable to eradicate the malignant progenitors and a significant portion of patients develop drug resistance after long-term use. STI571-based combined protocols seem to be feasible in the treatment of these patients. Yin et al[128] investigated the combined effects of imatinib and As4S4 on BCR-ABL and CML cells. They found synergic effects of STI-571 and As4S4 in the treatment of K562 cells and fresh CD34+ cells harvested from CML patients. Examination of cell cycles showed that As4S4 induced G2/M arrest, whereas imatinib induced G1 arrest. Using a number of parameters, such as morphology, annexin V/propidium iodide (PI), mitochondrial transmembrane potential, caspase-3 activity and Fas/Fas-L, the synergistic effects were revealed on induction of cell apoptosis, largely through the mitochondrial pathway. The 2 drugs also exhibited a synergistic effect in targeting BCR-ABL protein. Although As4S4 triggered its degradation and imatinib inhibited its tyrosine kinase activity, combined use of the 2 drugs led to lower protein/enzymatic activity levels of BCR-ABL. The in vitro data strongly suggested a potential clinical application of imatinib and As4S4 in combination on CML. To understand the mechanisms underlying the synergistic action of these agents, Du et al[129] applied cDNA microarrays, a component plane presentation integrated self-organizing map (CPP-SOM), and methods of protein biochemistry to study cell apoptosis induced by imatinib mesylate, ATO and a combination of the 2 agents in the CML cell line K562. Numerous features with temporo-spatial relationships were revealed, indicating the coordinated regulation of molecular networks from various aspects of pro-apoptotic and apoptotic activities in CML. Imatinib mesylate appears to mainly induce the intrinsic pathway of cell apoptosis, whereas ATO induces the endoplasmic reticulum (ER) stress-mediated pathway of cell apoptosis, and a combination of the 2 agents seems to more effectively induce the intrinsic, extrinsic and ER stress-mediated pathways of cell apoptosis, which results in a more effective and efficient induction of programmed cell death in K562 cells. This finding also appears to be supported by data derived from bone marrow cells of 2 patients with CML, 1 in chronic phase and the other in blast-crisis phase of the disease.
Studies on chronic lymphoid leukemia
CLL is a clinically heterogeneous disease originating from B lymphocytes that may differ in activation, maturation state or cellular subgroup[130]. It is the most common form of leukemia in developed countries and mainly affects elderly individuals. CLL cells can avoid death by responding to signal molecules or may undergo apoptosis, followed by replacement from proliferating precursor cells. CLL is a low-grade B-lineage lymphoid malignancy that follows an extremely variable course, with survival ranging from months to decades. Treatment decision is based on stage and disease progression, with chemotherapy, purine analogs (fludarabine, pentostatin and cladribine) and Rituximab (an anti-CD20 mAb) as first-line treatment[131].
The heritability of B-cell CLL is relatively high; however, no predisposing mutation has been convincingly identified. Raval et al[132] reported that loss or reduced expression of death-associated protein kinase 1 (DAPK1) underlined cases of heritable predisposition to CLL and the majority of sporadic CLL. Epigenetic silencing of DAPK1 by promoter methylation occurred in almost all sporadic CLL cases. Furthermore, these researchers defined a disease haplotype, which segregated with the CLL phenotype in a large family. DAPK1 expression of the CLL allele was downregulated by 75% in germline cells as a result of increased HOXB7 binding. In the blood cells from affected family members, promoter methylation resulted in the additional loss of DAPK1 expression. Thus, reduced expression of DAPK1 could result from germline predisposition, as well as epigenetic or somatic events causing or contributing to the CLL phenotype[132].
miRNA constitute a novel, phylogenetically extensive family of small RNA (~22 nucleotides) with potential roles in temporal and tissue-specific gene regulation. miRNA alterations are involved in the initiation and progression of human cancer. Calin et al[133] showed that miR15 and miR16 were located at chromosome 13q14, a region deleted in more than half of B-CLL. Detailed deletion and expression analysis showed that miR15 and miR16 were located within a 30-kb region of loss in CLL, and that both genes were deleted or downregulated in the majority (~68%) of CLL cases. Two years later Calin et al[134] reported genome-wide expression profiling of miRNA in human B-CLL using a microarray containing hundreds of human precursor and mature miRNA oligonucleotide probes. This approach allowed them to identify significant differences in miRNome expression between CLL samples and normal CD5+ B cells; data were confirmed by Northern blot analyses and real-time RT-PCR. At least two distinct clusters of CLL samples could be identified that were associated with the presence or absence of Zap-70 expression, a predictor of early disease progression. Two miRNA signatures were associated with the presence or absence of mutations in the expressed Ig variable-region genes or with deletions at 13q14, respectively. These data suggest that miRNA expression patterns have relevance to the biological and clinical behavior of CLL. In 2005, Calin et al[135] investigated whether miRNA profiles were associated with known prognostic factors in CLL. They evaluated the miRNA expression profiles of 94 samples of CLL cells for which the level of expression of ZAP-70, the mutational status of the rearranged immunoglobulin heavy-chain variable-region (IgVH) gene, and the time from diagnosis to initial treatment were known. They also investigated the genomic sequence of 42 miRNA genes to identify abnormalities. A unique miRNA expression signature composed of 13 genes (of 190 analyzed) differentiated cases of CLL with low levels of ZAP-70 expression from those with high levels and cases with unmutated IgVH from those with mutated IgVH . The same miRNA signature was also associated with the presence or absence of disease progression. A germ-line mutation in the miR-16-1–miR-15a primary precursor was also investigated, which caused low levels of miRNA expression in vitro and in vivo and was associated with deletion of the normal allele. Germ-line or somatic mutations were found in 5 of 42 sequenced miRNA in 11 of 75 patients with CLL, but no such mutations were found in 160 subjects without cancer (P<0.001). These data suggest that a unique miRNA signature is associated with prognostic factors and disease progression in CLL. Mutations in microRNA transcripts are common and may have functional importance.
Conclusion and perspectives
The pathogenesis of leukemia involves an array of molecular alterations that disrupt almost every facet of cell transformation. A large variety of genetic alterations, including point mutations, amplification, insertion, deletions, trisomy and chromosomal translocations, are important in leukemia initiation. Elucidation of genomic events and the cascade of their effects in cell function is crucial for identifying distinct subsets of leukemia and for developing new therapeutic strategies[136]. Two paradigms come from the identification of the key roles that PML-RARα and BCR-ABL fusion proteins play in APL and CML leukemogenesis, and from the development of ATRA/ATO and Imatinib, which greatly improve the prognosis of patients with APL or CML, respectively[137]. Optimal use of antileukemic agents has also resulted in a steady improvement in the treatment outcome of ALL[24]. Indeed, genomic medicine and the era of molecular targeted leukemia therapy have clearly arrived, and we should put the genome and targeted therapeutics into the doctors’ bag so that we can help patients to conquer leukemia, a notorious enemy of mankind, especially children.
References
- Parkin DM, Bray F, Ferlay J, Pisani P. Global Cancer Statistics, 2002. CA Cancer J Clin 2005;55:74-108.
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med 1985;103:620-5.
- Licht JD, Sternberg DW. The molecular pathology of acute myeloid leukemia. Hematology 2005; 2005: 137–42.
- Pui CH, Jeha S. New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat Rev Drug Discov 2007;6:149-65.
- Zhou GB, Zhao WL, Wang ZY, Chen SJ, Chen Z. Retinoic acid and arsenic for treating acute promyelocytic leukemia. PLoS Med 2005;2:33-8.
- Chen SJ, Zhu YJ, Tong JH, Dong S, Huang W, Chen Y, et al. Rearrangements in the second intron of the RARA gene are present in a large majority of patients with acute promyelocytic leukemia and are used as molecular marker for retinoic acid-induced leukemic cell differentiation. Blood 1991;78:2696-701.
- Wang YY, Zhou GB, Yin T, Chen B, Shi JY, Liang WX, et al. AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: Implication in stepwise leukemogenesis and response to Gleevec. Proc Natl Acad Sci USA 2005;102:1104-9.
- Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati T, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 2005;352:254-66.
- Gebhard C, Schwarzfischer L, Pham TH, Schilling E, Klug M, Andreesen R, et al. Genome-wide profiling of CpG methylation identifies novel targets of aberrant hypermethylation in myeloid leukemia. Cancer Res 2006;66:6118-28.
- Mrozek K, Bloomfield CD. Chromosome aberrations, gene mutations and expression changes, and prognosis in adult acute myeloid leukemia. Hematology 2006; 2006: 169–77.
- Zhou GB, Kang H, Wang L, Gao L, Liu P, Xie J, et al. Oridonin, a diterpenoid extracted from medicinal herbs, targets AML1-ETO fusion protein and shows potent antitumor activity with low adverse effects on t(8; 21) leukemia in vitro and in vivo. Blood 2007;109:3441-50.
- Wang L, Zhao WL, Yan JS, Liu P, Sun HP, Zhou GB, et al. Eriocalyxin B induces apoptosis of t(8; 21) leukemia cells through NF-kappaB and MAPK signaling pathways and triggers degradation of AML1-ETO oncoprotein in a caspase-3-dependent manner. Cell Death Differ 2007;14:306-17.
- Yang G, Thompson MA, Brandt SJ, Hiebert SW. Histone deacetylase inhibitors induce the degradation of the t(8; 21) fusion oncoprotein. Oncogene 2007;26:91-101.
- Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988;72:567-72.
- Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood 1996;88:1052-61.
- Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood 1997;89:3345-53.
- Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y, et al. A previously unidentified alternatively spliced isoform of t(8; 21) transcript promotes leukemogenesis. Nat Med 2006;12:945-9.
- Sobulo OM, Borrow J, Tomek R, Reshmi S, Harden A, Schlegelberger B, et al. MLL is fused to CBP, a histone acetyltrans-ferase, in therapy-related acute myeloid leukemia with at(11; 16)(q23; p13.3). Proc Natl Acad Sci USA 1997;94:8732-7.
- Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 1992;71:691-700.
- Thirman MJ, Levitan DA, Kobayashi H, Simon MC, Rowley JD. Cloning of ELL, a gene that fuses to MLL in a t(11; 19)(q23; p13.1) in acute myeloid leukemia. Proc Natl Acad Sci USA 1994;91:12110-4.
- Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 2006;354:2531-41.
- Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, et al. Nilotinib in Imatinib-Resistant CML and Philadelphia Chromosome-Positive ALL. N Engl J Med 2006;354:2542-51.
- Carroll WL, Bhojwani D, Min DJ, Raetz E, Relling M, Davies S, Pediatric acute lymphoblastic leukemia. Hematology 2003; 2003: 102–31.
- Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med 2006;354:166-78.
- Kau TR, Way JC, Silver PA. Nuclear transport and cancer: from mechanism to intervention. Nat Rev Cancer 2004;4:106-17.
- Chen Z, Chen SJ, Tong JH, Zhu YJ, Huang ME, Wang WC, et al. The retinoic acid alpha receptor gene is frequently disrupted in its 5' part in Chinese patients with acute promyelocytic leukemia. Leukemia 1991;5:288-92.
- Tong JH, Dong S, Geng JP, Huang W, Wang ZY, Sun GL, et al. Molecular rearrangements of the MYL gene in acute promyelocytic leukemia (APL, M3) define a breakpoint cluster region as well as some molecular variants. Oncogene 1992;7:311-6.
- Chen Z, Chen SJ. RARalpha and PML genes in acute promyelo-cytic leukemia. Leuk Lymphoma 1992;8:253-60.
- Qiang B. Human genome research in China. J Mol Med 2004;82:214-22.
- de TH. Chomienne C, Lanotte M, Degos L, Dejean A. The t(15; 17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990;347:558-61.
- Kakizuka A, Miller WH Jr, Umesono K, Warrell RP, Frankel SR, Murty VV, et al. Chromosomal translocation t(15; 17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991;66:663-74.
- Chen SJ, Chen Z, Chen A, Tong JH, Dong S, Wang ZY, et al. Occurrence of distinct PML-RAR-alpha fusion gene isoforms in patients with acute promyelocytic leukemia detected by reverse transcriptase/polymerase chain reaction. Oncogene 1992;7:1223-32.
- Geng JP, Tong JH, Dong S, Wang ZY, Chen SJ, Chen Z, et al. Localization of the chromosome 15 breakpoints and expression of multiple PML-RAR alpha transcripts in acute promyelocytic leukemia: a study of 28 Chinese patients. Leukemia 1993;7:20-6.
- Gu BW, Hu J, Xu L, Yan H, Jin WR, Zhu YM, et al. Feasibility and clinical significance of real-time quantitative RT-PCR assay of PML-RARalpha fusion transcript in patients with acute promyelocytic leukemia. Hematol J 2001;2:330-40.
- Gu BW, Xiong H, Zhou Y, Chen B, Wang L, Dong S, et al. Variant-type PML-RAR(alpha) fusion transcript in acute promyelocytic leukemia: use of a cryptic coding sequence from intron 2 of the RAR(alpha) gene and identification of a new clinical subtype resistant to retinoic acid therapy. Proc Natl Acad Sci USA 2002;99:7640-5.
- Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 1999;93:3167-215.
- Xu L, Zhao WL, Xiong SM, Su XY, Zhao M, Wang C, et al. Molecular cytogenetic characterization and clinical relevance of additional, complex and/or variant chromosome abnormalities in acute promyelocytic leukemia. Leukemia 2001;15:1359-68.
- Chen SJ, Zelent A, Tong JH, Yu HQ, Wang ZY, Derre J, et al. Rearrangements of the retinoic acid receptor alpha and promyelocytic leukemia zinc finger genes resulting from t(11; 17)(q23; q21) in a patient with acute promyelocytic leukemia. J Clin Invest 1993;91:2260-7.
- Chen Z, Brand NJ, Chen A, Chen S J, Tong JH, Wang ZY, et al. Fusion between a novel Kruppel-like zinc finger gene and the retinoic acid receptor-alpha locus due to a variant t(11; 17) translocation associated with acute promyelocytic leukaemia. EMBO J 1993;12:1161-7.
- Zhang T, Xiong H, Kan LX, Zhang CK, Jiao XF, Fu G, et al. Genomic sequence, structural organization, molecular evolution, and aberrant rearrangement of promyelocytic leukemia zinc finger gene. Proc Natl Acad Sci USA 1999;96:11422-7.
- Chen Z, Guidez F, Rousselot P, et al. PLZF-RAR alpha fusion proteins generated from the variant t(11; 17)(q23; q21) translocation in acute promyelocytic leukemia inhibit ligand-dependent transactivation of wild-type retinoic acid receptors. Proc Natl Acad Sci USA 1994;91:1178-82.
- Zhou GB, Chen SJ, Chen Z. Acute promyelocytic leukemia: a model of molecular target based therapy. Hematology 2005;10 Suppl 1:270-80.
- Grisolano JL, Wesselschmidt RL, Pelicci PG, Ley TJ. Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood 1997;89:376-87.
- Cheng GX, Zhu XH, Men XQ, Wang L, Huang QH, Jin XL, et al. Distinct leukemia phenotypes in transgenic mice and different corepressor interactions generated by promyelocytic leukemia variant fusion genes PLZF-RARalpha and NPM-RARalpha. Proc Natl Acad Sci USA 1999;96:6318-23.
- Chen LJ, Dong Y, Chen SY, Zhang L, Zhou GB, Chen B, et al. hCG-PLZF-RARalpha/hCG-RARalpha-PLZF transgenic mice developing into leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2005;13:924-31. Chinese..
- Niu C, Yan H, Yu T, Sun HP, Liu JX, Li XS, et al. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood 1999;94:3315-24.
- Zhang P, Wang SY, Hu LH. Arsenic trioxide treated 72 cases of acute promyelocytic leukemia. Chin J Hematol 1995;17:58-62.
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997;89:3354-60.
- Sun HD, Ma L, Hu XC, Zhang TD. Ai-Lin I treated 32 cases of acute promyelocytic leukemia. Chin J Integrat Chin West Med 1992;12:170-1.
- Fang J, Chen SJ, Tong JH, Wang ZG, Chen GQ, Chen Z, et al. Treatment of acute promyelocytic leukemia with ATRA and As2O3: a model of molecular target-based cancer therapy. Cancer Biol Ther 2002;1:614-20.
- Zhang JW, Wang JY, Chen SJ, Chen Z. Mechanisms of all-trans retinoic acid-induced differentiation of acute promyelocytic leukemia cells. J Biosci 2000;25:275-84.
- Zhu J, Chen Z, Lallemand-Breitenbach V, de The H. How acute promyelocytic leukaemia revived arsenic. Nat Rev Cancer 2002;2:705-13.
- Zhou G, Zhang J, Wang Z, Chen S, Chen Z. Treatment of acute promyelocytic leukaemia with all-trans retinoic acid and arsenic trioxide: a paradigm of synergistic molecular targeting therapy. Phil Trans R Soc B 2007;362:959-71.
- Chen Z, Chen GQ, Shen ZX, Chen SJ, Wang ZY. Treatment of acute promyelocytic leukemia with arsenic compounds: in vitro and in vivo studies. Semin Hematol 2001;38:26-36.
- Chen Z, Tong JH, Dong S, Zhu J, Wang ZY, Chen SJ, et al. Retinoic acid regulatory pathways, chromosomal translocations, and acute promyelocytic leukemia. Genes Chromosomes Cancer 1996;15:147-56.
- Chen Z, Wang ZY, Chen SJ. Acute promyelocytic leukemia: cellular and molecular basis of differentiation and apoptosis. Pharmacol Ther 1997;76:141-9.
- Nervi C, Ferrara FF, Fanelli M, Rippo MR, Tomassini B, Ferrucci PF, et al. Caspases mediate retinoic acid-induced degradation of the acute promyelocytic leukemia PML/RARalpha fusion protein. Blood 1998;92:2244-51.
- Brown D, Kogan S, Lagasse E, Weissman I, Alcalay M, Pelicci PG, et al. A PMLRARalpha transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci USA 1997;94:2551-6.
- vom BE, Zechel C, Heery D, Heine MJ, Garnier JM, Vivat V, Differential ligand-dependent interactions between the AF-2 activating domain of nuclear receptors and the putative transcriptional intermediary factors mSUG1 and TIF1. EMBO J 1996; 15: 110–24.
- Kamashev D, Vitoux D. de The H. PML-RARalpha-RXR oligomers mediate retinoid and rexinoid/cAMP cross-talk in acute promyelocytic leukemia cell differentiation. J Exp Med 2004;199:1163-74.
- Garzon R, Pichiorri F, Palumbo T, Visentini M, Aqeilan R, Cimmino A, et al. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene 2007;26:4148-57.
- Rocca B, Morosetti R, Habib A, Maggiano N, Zassadowski F, Ciabattoni G, et al. Cyclooxygenase-1, but not -2, is upregulated in NB4 leukemic cells and human primary promyelocytic blasts during differentiation. Leukemia 2004;18:1373-9.
- Kini AR, Peterson LA, Tallman MS, Lingen MW. Angiogenesis in acute promyelocytic leukemia: induction by vascular endothelial growth factor and inhibition by all-trans retinoic acid. Blood 2001;97:3919-24.
- Zhu J, Guo WM, Yao YY, Zhao WL, Pan L, Cai X, et al. Tissue factors on acute promyelocytic leukemia and endothelial cells are differently regulated by retinoic acid, arsenic trioxide and chemotherapeutic agents. Leukemia 1999;13:1062-70.
- Xiao S, Li D, Zhu HQ, Song MG, Pan XR, Jia PM, et al. RIG-G as a key mediator of the antiproliferative activity of interferon-related pathways through enhancing p21 and p27 proteins. Proc Natl Acad Sci USA 2006;103:16448-53.
- Mao M, Yu M, Tong JH, Ye J, Zhu J, Huang QH, et al. RIG-E, a human homolog of the murine Ly-6 family, is induced by retinoic acid during the differentiation of acute promyelocytic leukemia cell. Proc Natl Acad Sci USA 1996;93:5910-4.
- Yu M, Tong JH, Mao M, Kan LX, Liu MM, Sun YW, et al. Cloning of a gene (RIG-G) associated with retinoic acid-induced differentiation of acute promyelocytic leukemia cells and representing a new member of a family of interferon-stimulated genes. Proc Natl Acad Sci USA 1997;94:7406-11.
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 2004;5:730-7.
- Liu TX, Zhang JW, Tao J, Zhang RB, Zhang QH, Zhao CJ, et al. Gene expression networks underlying retinoic acid-induced differentiation of acute promyelocytic leukemia cells. Blood 2000;96:1496-504.
- Zhu J, Koken MH, Quignon F, Chelbi-Alix MK, Degos L, Wang ZY, et al. Arsenic-induced PML targeting onto nuclear bodies: implications for the treatment of acute promyelocytic leukemia. Proc Natl Acad Sci USA 1997;94:3978-83.
- Lallemand-Breitenbach V, Zhu J, Puvion F, Koken M, Honoré N, Doubeikovsky A, et al. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J Exp Med 2001;193:1361-71.
- Muller S, Matunis MJ, Dejean A. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J 1998;17:61-70.
- Hayakawa F, Privalsky ML. Phosphorylation of PML by mitogen-activated protein kinases plays a key role in arsenic trioxide-mediated apoptosis. Cancer Cell 2004;5:389-401.
- Zhu J, Lallemand-Breitenbach V. de TH. Pathways of retinoic acid- or arsenic trioxide-induced PML/RARalpha catabolism, role of oncogene degradation in disease remission. Oncogene 2001;20:7257-65.
- Shen ZX, Shi ZZ, Fang J, Gu BW, Li JM, Zhu YM, et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA 2004;101:5328-35.
- Liu YF, Zhu YM, Shen SH, Shen ZX, Li JM, Chen SJ, et al. Molecular response in acute promyelocytic leukemia: a direct comparison of regular and real-time RT-PCR. Leukemia 2006;20:1393-9.
- Zheng PZ, Wang KK, Zhang QY, Huang QH, Du YZ, Zhang QH, et al. Systems analysis of transcriptome and proteome in retinoic acid/arsenic trioxide-induced cell differentiation/apoptosis of promyelocytic leukemia. Proc Natl Acad Sci USA 2005;102:7653-8.
- Leung J, Pang A, Yuen WH, Kwong YL, Tse EWC. Relationship of expression of aquaglyceroporin 9 with arsenic uptake and sensitivity in leukemia cells. Blood 2007;109:740-6.
- Verschuus AC. Acute myeloblastic leukemia with maturation. Orphanet Encyclopedia. May 2004; : 1–5.http: //www.orpha.net/data/patho/GB/uk-AMLM2.pdf
- Licht JD. AML1 and the AML1-ETO fusion protein in the pathogenesis of t(8; 21) AML. Oncogene 2001;20:5660-79.
- Rubnitz JE, Raimondi SC, Halbert AR, Tong X, Srivastava DK, Razzouk I, et al. Characteristics and outcome of t(8; 21)-positive childhood acute myeloid leukemia: a single institution’s experience. Leukemia 2002;16:2072-7.
- Peterson LF, Zhang DE. The 8; 21 translocation in leukemo-genesis. Oncogene 2004;23:4255-62.
- Amann JM, Nip J, Strom DK, Lutterbach B, Harada H, Lenny N, et al. ETO, a target of t(8; 21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol 2001;21:6470-83.
- Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. ETO, fusion partner in t(8; 21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci USA 1998;95:10860-5.
- Uchida H, Zhang J, Nimer SD. AML1A and AML1B can transactivate the human IL-3 promoter. J Immunol 1997;158:2251-8.
- Takahashi A, Satake M, Yamaguchi-Iwai Y, Bae SC, Lu J, Maruyama M, et al. Positive and negative regulation of granulocyte-macrophage colony-stimulating factor promoter activity by AML1-related transcription factor, PEBP2. Blood 1995;86:607-16.
- Frank R, Zhang J, Uchida H, Meyers S, Hiebert SW, Nimer SD. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML1B. Oncogene 1995;11:2667-74.
- Klampfer L, Zhang J, Zelenetz AO, Uchida H, Nimer SD. The AML1/ETO fusion protein activates transcription of BCL-2. Proc Natl Acad Sci USA 1996;93:14059-64.
- Shimizu K, Kitabayashi I, Kamada N, Abe T, Maseki N, Suzukawa K, et al. AML1-MTG8 leukemic protein induces the expression of granulocyte colony-stimulating factor (G-CSF) receptor through the up-regulation of CCAAT/enhancer binding protein epsilon. Blood 2000;96:288-96.
- Higuchi M, O’Brien D, Kumaravelu P, Lenny N, Yeoh EJ, Downing JR. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8; 21) acute myeloid leukemia. Cancer Cell 2002;1:63-74.
- Okuda T, Cai Z, Yang S, Lenny N, Lyu CJ, van Deursen JM, et al. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood 1998;91:3134-43.
- Yuan Y, Zhou L, Miyamoto T, Iwasaki H, Harakawa N, Hetherington CJ, et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc Natl Acad Sci USA 2001;98:10398-403.
- Fenske TS, Pengue G, Mathews V, Hanson PT, Hamm SE, Riaz N, et al. Stem cell expression of the AML1/ETO fusion protein induces a myeloproliferative disorder in mice. Proc Natl Acad Sci USA 2004;101:15184-9.
- de Guzman CG, Warren AJ, Zhang Z, Gartland L, Erickson P, Drabkin H, et al. Hematopoietic stem cell expansion and distinct myeloid developmental abnormalities in a murine model of the AML1-ETO translocation. Mol Cell Biol 2002;22:5506-17.
- Yan M, Burel SA, Peterson LF, Kanbe E, Iwasaki H, Boyapati A, et al. From the Cover: Deletion of an AML1-ETO C-terminal NcoR/SMRT-interacting region strongly induces leukemia development. Proc Natl Acad Sci USA 2004;101:17186-91.
- Schessl C, Rawat VPS, Cusan M, Kanbe E, Iwasaki H, Boyapati A, et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J Clin Invest 2005;115:2159-68.
- Buchholz F, Refaeli Y, Trumpp A, Bishop JM. Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse. EMBO Rep 2000;1:133-9.
- Rhoades KL, Hetherington CJ, Harakawa N, Yergeau DA, Zhou L, Liu LQ, et al. Analysis of the role of AML1-ETO in leukemo-genesis, using an inducible transgenic mouse model. Blood 2000;96:2108-15.
- Mulloy JC, Cammenga J, Berguido FJ, Wu K, Zhou P, Comenzo RL, et al. Maintaining the self-renewal and differentiation potential of human CD34+ hematopoietic cells using a single genetic element. Blood 2003;102:4369-76.
- Grisolano JL, O’Neal J, Cain J, Tomasson MH. An activated receptor tyrosine kinase, TEL/PDGFbetaR, cooperates with AML1/ETO to induce acute myeloid leukemia in mice. Proc Natl Acad Sci USA 2003;100:9506-11.
- Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood 1998;92:2322-33.
- Billstrom R, Johansson B, Fioretos T, Garwicz S, Malm C, Zettervall O, et al. Poor survival in t(8; 21) (q22; q22)-associated acute myeloid leukaemia with leukocytosis. Eur J Haematol 1997;59:47-52.
- Leblanc T, Berger R. Molecular cytogenetics of childhood acute myelogenous leukaemias. Eur J Haematol 1997;59:1-13.
- Lee KW, Choi IS, Roh EY, Kim DY, Yun T, Lee DS, et al. Adult patients with t(8; 21) acute myeloid leukemia had no superior treatment outcome to those without t(8; 21): a single institution’s experience. Ann Hematol 2004;83:218-24.
- Matsumoto Y, Mori M, Ohtsuki T, Muroi K, Hatake K, Komatsu N, et al. Outcome of acute myelogenous leukemia in 41 patients treated with idarubicin: the prognosis of t(8; 21) cases. Rinsho Ketsueki 2001;42:15-22. Japanese..
- Cho EK, Bang SM, Ahn JY, Yoo SM, Park PW, Seo YH, et al. Prognostic value of AML 1/ETO fusion transcripts in patients with acute myelogenous leukemia. Korean J Intern Med 2003;18:13-20.
- Shigesada K. van de SB, Liu PP. Mechanism of leukemogenesis by the inv(16) chimeric gene CBFB/PEBP2B-MHY11. Oncogene 2004;23:4297-307.
- Helbling D, Mueller BU, Timchenko NA, Schardt J, Eyer M, Betts DR, et al. CBFB-SMMHC is correlated with increased calreticulin expression and suppresses the granulocytic differentiation factor CEBPA in AML with inv(16). Blood 2005;106:1369-75.
- Wunderlich M, Krejci O, Wei J, Mulloy JC. Human CD34+ cells expressing the inv(16) fusion protein exhibit a myelomonocytic phenotype with greatly enhanced proliferative ability. Blood 2006;108:1690-7.
- Pession A, Martino V, Tonelli R, Kersey JH, Nakamura T, Canaani E, et al. MLL-AF9 oncogene expression affects cell growth but not terminal differentiation and is downregulated during monocyte-macrophage maturation in AML-M5 THP-1 cells. Oncogene 2003;22:8671-6.
- Barabe F, Kennedy JA, Hope KJ, Dick JE. Modeling the initiation and progression of human acute leukemia in mice. Science 2007;316:600-4.
- Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006;10:257-68.
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006;442:818-22.
- Liu TX, Becker MW, Jelinek J, Wu WS, Deng M, Mikhalkevich N, et al. Chromosome 5q deletion and epigenetic suppression of the gene encoding [alpha]-catenin (CTNNA1) in myeloid cell transformation. Nat Med 2007;13:78-83.
- Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007;446:758-64.
- Zhu YM, Zhao WL, Fu JF, Shi JY, Pan Q, Hu J, et al. NOTCH1 mutations in T-cell acute lymphoblastic leukemia: prognostic significance and implication in multifactorial leukemogenesis. Clin Cancer Res 2006;12:3043-9.
- Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005;8:407-19.
- Chen SJ, Wang Q, Tong JH, Xie WJ, Cao Q, Cai JR, et al. Monoallelic deletions of the P53 gene in Chinese patients with chronic myelogenous leukemia in blastic crisis. Nouv Rev Fr Hematol 1991;33:481-4.
- Su XY, Wong N, Cao Q, Yu LZ, Niu C, Wickham N, et al. Chromosomal aberrations during progression of chronic myeloid leukemia identified by cytogenetic and molecular cytogenetic tools: implication of 1q12-21. Cancer Genet Cytogenet 1999;108:6-12.
- Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer 2005;5:172-83.
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344:1031-7.
- O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003;348:994-1004.
- Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006;355:2408-17.
- Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001;344:1038-42.
- Talpaz M, Silver RT, Druker BJ, Goldman JM, Gambacorti-Passerini C, Guilhot F, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood 2002;99:1928-37.
- Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 2002;346:645-52.
- Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood 2002;99:3530-9.
- Yin T, Wu YL, Sun HP, Sun GL, Du YZ, Wang KK, et al. Combined effects of As4S4 and imatinib on chronic myeloid leukemia cells and BCR-ABL oncoprotein. Blood 2004;104:4219-25.
- Du Y, Wang K, Fang H, Li J, Xiao D, Zheng P, et al. Coordination of intrinsic, extrinsic, and endoplasmic reticulum-mediated apoptosis by imatinib mesylate combined with arsenic trioxide in chronic myeloid leukemia. Blood 2006;107:1582-90.
- Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med 2005;352:804-15.
- Hallek M. Chronic lymphocytic leukemia (CLL): First-line treatment. Hematology Am Soc Hematol Edu Program 2005; 285–91.
- Raval A, Tanner SM, Byrd JC, Angerman EB, Perko JD, Chen SS, et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007;129:879-90.
- Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 2002;99:15524-9.
- Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA 2004;101:11755-60.
- Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 2005;353:1793-801.
- Mecucci C, Rosati R, Starza RL. Genetic profile of acute myeloid leukemia. Rev Clin Exp Hematol 2002;6:3-25.
- Hu J, Zhou GB, Wang ZY, Chen SJ, Chen Z. Mutant transcription factors and tyrosine kinases as therapeutic targets for leukemias: from acute promyelocytic leukemia to chronic myeloid leukemia and beyond. Adv Cancer Res 2007;98:191-220.