
Cancer chemoprevention by phytochemicals: potential molecular targets, biomarkers and animal models1
Introduction
Chemoprevention is an active cancer preventive strategy to inhibit, delay or reverse human carcinogenesis, using naturally occurring or synthetic chemical agents. The term “chemoprevention” was first introduced by Dr Michael B. Sporn, when he referred to the prevention of cancer development by natural forms of vitamin A and by its synthetic analogs[1]. Thereafter, a variety of naturally-occurring dietary compounds have been shown to possess significant chemopreventive effects and many experimental attempts have been made to address their underlying mechanisms of actions[2]. Numerous cancer cell lines and animal cancer models have been used to evaluate the chemopreventive effects of phytochemicals as well as to elucidate their mechanisms of cancer prevention. These studies have resulted in the discovery of several new phytochemicals that possess cancer preventive effects, such as isothiocyanates from cruciferous vegetables, polyphenols from green and black tea, and flavonoids from soybeans[2]. Several cellular mechanisms contribute to the overall cancer preventive effects of these dietary phytochemicals. These include oxidative or electrophilic stresses that can trigger a wide variety of cellular events such as increasing expression of detoxifying enzymes and/or antioxidant enzymes, inhibiting cell cycle progression and cell proliferation, inducing differentiation and apoptosis, inhibiting expression and functional activation of oncogenes, increasing expression of tumor-suppressor genes, and inhibiting angiogenesis and metastasis by modulating cellular signaling pathways[2,3]. These signal transduction pathways are now recognized as potential molecular targets for chemoprevention by dietary phytochemicals[4]. The scope of the present review will focus on the molecular basis of chemopreventive potential of dietary phytochemicals, with special emphasis on their effects on cellular signaling cascades mediated by nuclear factor E2-related factor 2 (Nrf2), nuclear factor-kappaB (NF-κB), cyclooxy-genases-2 (COX-2), activator protein-1 (AP-1), mitogen-activated protein kinases (MAPKs), and inflammatory mediator-related pathways, as well as several animal cancer models for cancer chemoprevention research.
Natural dietary phytochemicals
Over millions of years, plants have developed the capacity to synthesize a diverse array of chemicals. In general, these phytochemicals function to attract beneficial organisms, repel harmful organisms, serve as photoprotectants and respond to environmental cues for the plants’ survival. For example, numerous classes of phytochemicals, including flavonoids, polyphenols and anthocyanins function as phytoalexins, substances that assist a plant to resist patho-gens. Humans and animals have been ingesting plants since the inception of evolution. Needless to say both humans and animals have developed a robust digestive system that helps them reap the benefits of such ingestion. The primary research in our laboratory is focused towards understanding the beneficial effects of some of these dietary phyto-chemicals.
Studies from our laboratory, as well as others, have shown that these phytochemicals can act as blocking agents obstructing the initiation phase of carcinogenesis or suppressing agents retarding the promotion and progression phases of carcinogenesis. Recently, we showed that sulforaphane (SFN) a component of cruciferous vegetables inhibited adenoma formation in the gastro-intestinal tract of ApcMin/+mice[5,6]. Profiling the gene expression modulated by SFN in the small intestinal polyps of these mice revealed that several pro-apoptotic genes such as MBD4, TNF-7 and TNF (ligand)-11 were upregulated while pro-survival genes such as cyclin-D2, integrin-β1, Wnt-9A were found to be downregulated[7]. In MCF-7, a breast cancer cell line and MCF-10F, a non-cancerous human breast cell-line, SFN has been shown to inhibit DNA adduct formation by polycyclic aromatic hydrocarbon-benzo[a]pyrene (BaP) and 1,6 dinitro-pyrene[8]. Curcumin the diketone obtained from the Indian spice turmeric has been long known as an anti-inflammatory and anti-cancer agent. Curcumin has been shown to inhibit tumor initiation by BaP and 7,12-dimethyl-benz[a]anthracene (DMBA) and also to inhibit tumor promotion induced by 12-O-tetradecanoylphorbol-13-acetate (TPA)[9]. Phenylethyliso-thiocyanate (PEITC), another component of cruciferous vegetables has been shown to induce apoptosis in the human colon cancer cell line HT-29 by activating the mitochondrial caspase cascade[10,11]. It has been shown to potently inhibit NF-κB activity in human prostate cancer (PC3) cells by inhibiting IKKα/β signaling pathway[12]. Interestingly a combination of curcumin and PEITC demonstrated an additive effect in inducing apoptosis in PC3 cells by significantly inhibiting EGFR and its downstream signaling molecules PI3K and Akt[13]. A similar combination significantly inhibited tumor formation in athymic nude mice implanted with PC3 xenografts[14]. Transgenic adenocarcinoma of the mouse prostate (TRAMP) is a transgenic mouse model that recapitulates all of the salient features of human prostate cancer. Both curcumin and PEITC individually as well as in combination inhibited tumor development in these mice (unpubli-shed data). (-) epigallocatechin gallate (EGCG) a popular component of green tea has been shown to induce apoptosis in HT-29 cells by damaging the mitochondria and activating the caspases[15]. It has also been shown to promote apopto-sis in T24 human bladder cancer cells by modulating the PI3K/Akt signaling pathway and Bcl-2 family proteins[16].
It is important to note that though each of these phyto-chemicals is potent in inhibiting tumor and/or cancer development, they are also non-toxic to the normal cells. What really sets apart their differential effects in abnormal cancer cells versus normal cells is their ability to induce apoptotic pathways to impede cancer in abnormal cancer cells and at the same time manipulate levels of metabolizing enzymes and induce detoxifying enzymes rendering them non-toxic to normal cells. SFN has been shown to increase mRNA and protein levels of quinone reductase (QR), UDP-glucuronyltransferase (UGT) and glutathione-S-transferase (GST) and heme oxygenase (HO)-1 and this increase is mediated by the induction of ARE. More recently, work from our laboratory has demonstrated that the inhibition of p38 MAPK isoform by SFN contributes to the induction of ARE-mediated increases in HO-1 gene expression[17]. SFN has also been shown to decrease the mRNA levels of CYP3A4, which translates to a decrease in CYP3A4 activity[18]. Likewise PEITC, EGCG, and curcumin have also been shown to induce phase II detoxifying enzymes – UGT, GST, HO-1[19,20]. Thus it can be inferred that careful manipulation of multiple signaling cascades by dietary phytochemicals within the cell contribute to the pleiotropic effects of these potential chemo-preventive agents.
Nrf2 signaling pathways
Nrf2, a member of the Cap’n’collar family of basic region-leucine zipper (bZIP) transcription factors (Figure 1A) that can act as a master regulator of ARE-driven transactivation of antioxidant genes[21]. Six conserved domains of Nrf2 are designated Nrf2-epichlorohydrin (ECH) homology (Neh) 1–Neh 6. Neh1 corresponds to the CNC homology region and bZIP domain. The amino and carboxyl termini of the proteins are also highly conserved, and are referred to as Neh2 and Neh3, respectively. In addition, there are two conserved acidic domains (Neh4 and Neh5) as well as a serine-rich conserved region (Neh6).
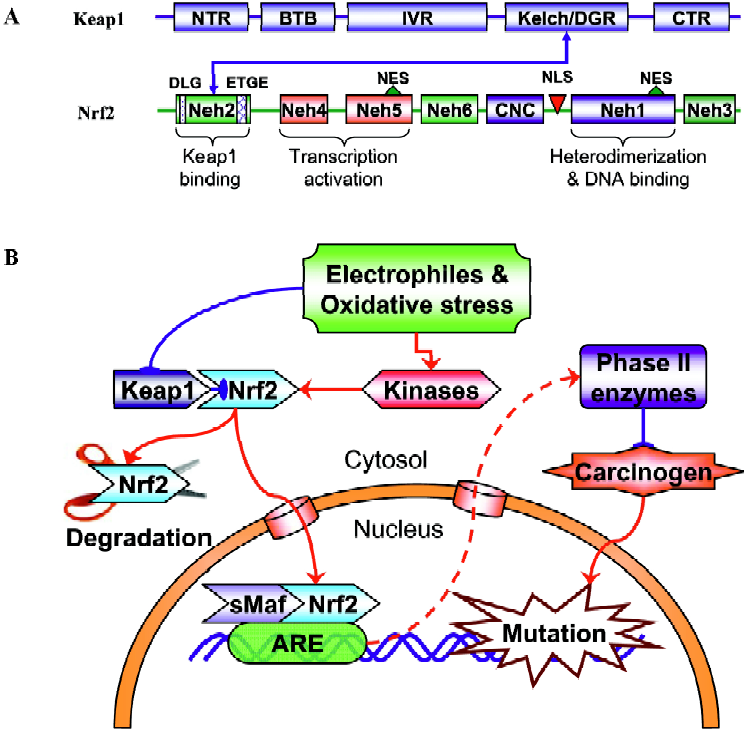
Its important role in regulating the expression of many mammalian detoxifying and antioxidant enzymes under oxidative or electrophilic stress has been verified in various Nrf2-deficient mice studies, in which the expression of these enzymes were dramatically abolished and the Nrf2 knockout mice were much more susceptible to carcinogen-induced carcinogenesis[22–24]. As described above, many phytochemi-cals exert their chemopreventive effects by blocking the initiation stage of cancer development. For a subset of these compounds, chemoprotection is mainly derived from the induction of Nrf2/ARE-regulated genes. For example, the phase II GST gene can be induced by phenolic antioxidant butylated hydroxyanisole (BHA) and ethoxyquin[25] or by isothiocyanate[26]. The redox-sensitive stress gene HO-1 can be induced by curcumin and caffeic acid phenethyl ester (CAPE) through an Nrf2 signaling pathway[27]. Under basal unstimulated conditions, Nrf2 is sequestered in the cytoplasm as an inactive complex with its cytosolic repressor Kelch-like ECH associated protein 1 (Keap1, Figure 1A). Nrf2 is tethered to Keap1 in the cytosol. Most of the Nrf2 proteins are targeted to proteosomal degradation presumably with low levels of recycling to the nucleus[28]. Dissociation of Nrf2 from the inhibitory protein Keap1 is a prerequisite for nuclear translocation and subsequent DNA binding of Nrf2[29]. Keap1 is a cysteine-rich protein that interacts with the ETGE motif within the N-terminal Neh2 domain of Nrf2[30] (Figure 1A). Many studies have shown that challenges with chemopreventive agents can lead to enhanced nuclear accumulation of Nrf2, therefore activating Nrf2-dependent gene transcription[24,25,27,31]. However, the exact mechanisms by which these phytochemicals or the generated exogenous and/or endogenous oxidative and/or electrophile stresses trigger the Nrf2 transactivation activity are still unclear. The binding of Keap1 to Nrf2 represses Nrf2-mediated gene transcription under homeostatic conditions. Upon exposure to above threshold levels of chemopreventive chemicals or oxidative stress, Nrf2 is able to escape Keap1-mediated repression, translocate to the nucleus, where Nrf2 binds to the ARE in association with small Maf proteins inside the nucleus, and activate expression of its target genes (Figure 1B). Chemopreventive compounds may also directly cause the cleavage of the disulfide bond between Nrf2 and Keap1. By inducing the phase II detoxifying and antioxidant enzyme genes, phytochemicals increase the detoxification of pro-carcinogens or carcinogens and protect normal cells from the damage of electrophiles and reactive oxygen intermedi-ates, thus decreasing the incidence of initiation and reducing the risk of cancer. Considering the great structural diversity of the inducers that regulate the Nrf2 signaling path-way[32], a mechanism of activation requiring the direct binding or interaction of phytochemicals that activate Nrf2 with a structurally complementary receptor appears to be quite unlikely. Therefore, many phytochemicals might regulate Nrf2-mediated gene transcription by different mechanisms. It is important to note that many chemopreventive agents could possess pro-oxidant properties, meaning they can generate oxidative and/or electrophilic stress by themselves in the cells. The chemical stress generated by chemopreven-tive agents appears to be dose-dependent, mild at low concentrations with sufficient signal strength to activate the cellular defense systems that lead to the coordinated activation of the Nrf2 signaling pathway. However, this low stress is believed to be at a subtoxic level that would not cause any adverse effects such as DNA damage, mutagenicity, or degeneration of tissues as traditionally caused by carcinogens.
NF-κB and AP-1 signaling pathways
Nuclear factor-kappaB NF-κB is an ubiquitous redox-sensitive transcription factor that includes six family members: NF-κB1 (p50 and its precursor p105), NF-κB2 (p52 and its precursor p100), RelA (p65), RelB (p68), c-Rel (p75) and v-Rel (Figure 2). These proteins bind to a specific promoter of target genes and regulate a wide range of cellular events, such as cell proliferation, cell cycle control, apoptosis, differentiation and tumorgenesis by forming homodimers or heterodimers with each other. In resting or unstimulated cells, NF-κB is sequestered in the cytoplasm by binding to IκBα (IκBα, IκBα, IκBα) that masks the nuclear localization sequence (NLS) in NF-κB. When cells receive the signals that activate NF-κB signaling pathways, IκB protein will be phosphorylated at Ser/Thr residues by the upstream IκB kinase (IKK) complex (consisting of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ or NEMO) and undergo ubiquitin-mediated proteosomal degrada-tion[33,34], which in turn enables the nuclear import machinery to recognize the NLS and transports NF-κB to the nucleus. It is reported that activation of NF-κB can promote or inhibit apoptosis, depending on the cell type and other condi-tions[35,36], but activation of NF-κB is generally thought to protect cells against apoptotic stimuli in most tumor cell lines, presumably via the induction of cell survival genes[37]. This notion agrees well with the fact that the NF-κB signaling pathway is constitutively activated in leukemia[38], prost-ate[39], breast[40] and pancreatic cancers[41] and that the blockade of NF-κB can increase the sensitivity of cancers to chemotherapeutic drugs[42]. Therefore, inhibition of the NF-κB signaling pathway has emerged as an important target of chemotherapeutic and chemopreventive compounds[42].
Activator protein-1 is another redox-sensitive transcription factor that can be regulated by a large variety of stimuli, including pro-inflammatory cytokines, growth factors, oxidative stress and tumor promoters[43] (Figure 2). AP-1 is a dimeric transcription factor that consists of the basic leucine-zipper family members, including the Jun (c-Jun, JunB and JunD), Fos (c-Fos, FosB, Fra1 and Fra2), ATF (ATF2, B-ATF, JDP1 and JDP2), and Maf (MafA, MafB, c-Maf and MafG/F/K) protein families, in which Jun and Fos dimer is the most common partner proteins found in the euka-ryotes[44,45]. When activated, AP-1 recognizes and binds to the TPA response element (TRE) or cAMP response element within the promoter region of target genes. Because of its implication in a wide variety of cellular responses, the role of AP-1 in carcinogenesis has been heavily investigated. Inhibition of Fos and Jun expression in mice fibroblasts and erythroleukaemia cells has indicated that AP-1 is required for cell proliferation and cell cycle progression[46]. Using mice overexpressing c-Fos, Wang et al showed a close relationship between c-Fos expression levels and chondrogenic tumor development[47]. c-Jun activity suppression, using a dominant-negative c-Jun in basal keratinocytes or conditional inactivation of c-Jun in the liver resulted in the interference of the development of chemically-induced papillomas and liver tumors, respectively[48,49]. MAPKs (ERK, JNK and p38 MAPK) are critical mediators of AP-1 activity in response to pro-inflammatory cytokines or exogenous stress[50]. Activation of JNK and p38 MAPK phosphorylates and activates c-Jun and ATF2 proteins, respectively, which in turn leads to the activation of AP-1 target genes. JNK can also cross-talk with PI3K to upregulate AP-1 activity[51].
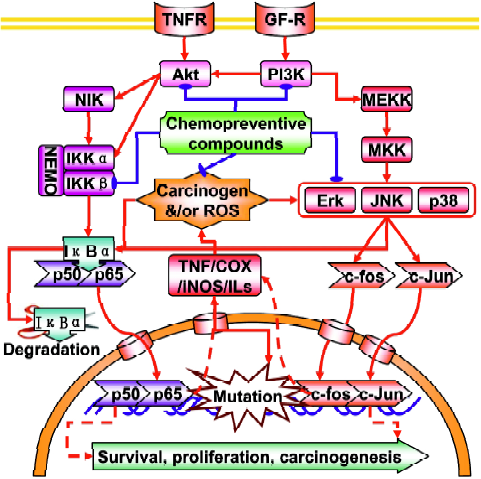
Given that the regulation of NF-κB and AP-1 by intracellular signaling cascades is complex, it is imperative to know how dietary chemopreventive compounds modulate the intracellular signaling cascades to affect the basal and/or inducible NF-κB and AP-1 activity. At present, a vast number of studies have reported the regulation of NF-κB and AP-1 activities by chemopreventive compounds. Due to limited space, however, we will focus on two well-known dietary chemopreventive compounds (isothiocyanates and curcumin) and discuss how they modulate NF-κB and AP-1 activities. Isothiocyanates (ITCs) are among a class of chemopreventive compounds, which abundantly exist in cruciferous vegetables such as broccoli, watercress, Brussels sprouts, cabbage and cauliflower. ITCs are characterized by the chemical structure of R-N=C=S, where R designates an alkyl or aryl group. We have previously shown that treatment of ITCs, such as SFN, PEITC and allyl isothiocyanate (AITC) significantly inhibited the lipopolysaccharide (LPS)-induced NF-κB-dependent gene expression in human colon carcinoma HT-29 cells, stably transfected with NF-κB-luciferase reporter and this event was closely correlated with the suppression of IκBα phosphorylation[52]. Similarly, we found that the exposure of SFN and PEITC to human prostate cancer PC-3 cells strongly suppressed NF-κB target gene expression by attenuating phosphorylation of upstream regulatory kinases, IKKα and IKKβ[12]. Likewise, the effects of a variety of ITCs on AP-1 activity have been intensively investigated. For example, Zhu et al have shown that SFN can inhibit UV-induced AP-1 activation in human keratinocytes[53]. In another study, we found that, while treatment of PEITC and SFN increased AP-1-dependent gene expression in HT-29 cells at selected concentrations, AITC increased AP-1-dependent gene expression in a dose-dependent manner[54]. Together, these results indicate that the regulation of NF-κB and AP-1 activity by ITCs is highly variable, depending on the chemical structures of ITCs, cell types and/or many other cellular factors.
MAPK signaling pathways
Mitogen-activated protein kinases, characterized as proline-directed serine/threonine (ProXSer/ThrPro) kina-ses[55,56], are important cellular signaling components that convert various extracellular signals into intracellular responses through serial phosphorylation cascades[57] (Figure 2). At the present time, three distinct but parallel MAP kinase cascades (ERK, JNK, and p38) have been identified in mammalian cells[58,59]. Each consists of a module of three kinases: a MAPK kinase kinase (MAPKKK), which phosphorylates and activates a MAPK kinase (MAPKK), which, in turn, phosphorylates and activates a MAPK. Although the MAPK, in particular the ERK, has been known to be activated by mitogens and growth factors, many environmental stress stimuli such as UV and ionizing radiation can also activate JNK and p38 MAPK, with the consequences of apoptotic cell death[60]. Many chemotherapeutic drugs have been known to activate JNK and p38 MAPK and their activation had been implicated in apoptosis[61]. Once activated, these three MAPKs (ERK, JNK, and p38) can phosphorylate many transcription factors, such as c-Myc, p62TCF/Elk-1, c-Jun, ATF2, CHOP/GADD153, MEF2C, and SAP-1, and ultimately leading to the changes in gene expression[62]. Given the fact that MAPKs are activated by such a wide range of factors, these signaling cascades may serve as a common mechanism and integrate with other signaling pathways to control cellular responses to various extracellular stimuli, including xenobiotics and pharmacological agents. The ability of curcumin to regulate the MAPKs signaling pathway might contribute to the suppression of inflammation by curcumin. Lee et al have reported that curcumin inhibits phorbol ester-induced upregulation of COX-2 and matrix metalloproteinase-9 by blocking ERK1/2 phosphorylation and NF-κB transcriptional activity in MCF10A human breast epithelial cells[63]. Chen et al found that curcumin inhibits JNK activation induced by various agonists including PMA plus ionomycin, anisomycin, UV-C, and γ-radiation[64]. Salh et al reported that curcumin is able to attenuate dinitrobenzene sulfonic acid (DNB)-induced experimental colitis through a reduction in the activity of p38 MAPK[65]. In addition, Li et al have reported that indole-3-carbinol (I3C) and its active metabolite 3,3"-diindolylmethane (DIM) down-regulate the expression of MAP2K3, MAP2K4, MAP4K3 and MARK3 in PC3 prostate cancer cells. The results suggest that I3C and DIM suppress MAPK signaling[66].
Inflammatory mediator-related pathways
Tumor necrosis factor-α (TNF-α), is a pro-inflammatory cytokine with a wide variety of biological functions in inflammation like tissue remodeling, alteration of epithelial barrier permeability, increasing vascular permeability, activation of macrophages, recruitment of inflammatory cells, and upregulation of cell adhesion molecules[67]. TNF-α can be produced and released by activated monocytes, macro-phages, T cells, and mast cells. The induction of pro-inflammatory genes by TNF has been linked to most diseases. Almost all cell types, when exposed to TNF, activate NF-κB, leading to the expression of inflammatory genes. These include pro-inflammatory cytokines, chemokines, COX-2, inducible nitric oxide synthase (iNOS), lipoxygenase-2 (LOX-2), and cell adhesion molecules. TNF has been found to be a growth factor for most tumor cells[68]. These include ovarian cancer cells, cutaneous T cell lymphoma[69], acute myelogenous leukemia[70], and B cell lymphoma[71]. Because of the critical role of TNF in mediating tumorigenesis, agents that can suppress TNF activity have potential for the therapy of TNF-linked diseases. Moore et al reported that TNF-α knockout mice have been shown to be resistant to skin carcinogenesis[72], suggesting that neutralization of TNF-α production may be useful in cancer treatment and prevention. Phytochemicals such as curcumin[73], green tea polyphenols EGCG[74] and resveratrol[75] have been shown to suppress TNF production.
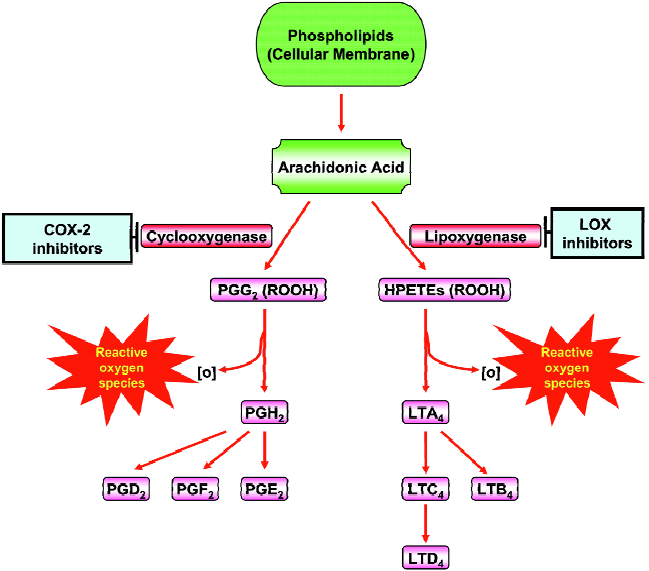
Arachidonic acid (AA) metabolism diverges into the COX and the LOX pathways (Figure 3). The COX pathway leads to prostaglandin (PG) and thromboxane production and the LOX pathway leads to the leukotrienes (LTs) and hydro-peroxyeicosatetraenoic acids (HPETEs). These classes of inflammatory molecules exert profound biological effects that enhance the development and progression of human cancers. COX-1 and 2 are the rate-limiting enzymes in the conversion of AA to PGs (Figure 3). The two COX isoforms of PGH synthase have distinct tissue distributions and physiological functions. COX-1 is constitutively expressed in many tissues and cell types, whereas the inducible isoenzyme COX-2 is pro-inflammatory in nature and expressed only in response to certain stimuli such as mitogens, cytokines, growth factors, or hormones. COX-2 is overexpressed in practically every pre-malignant and malignant condition involving the colon, liver, pancreas, breast, lung, bladder, skin, stomach, head and neck[76]. Depending on the stimulus and the cell type, several transcription factors including AP-1, NF-IL-6, and NF-κB can stimulate COX-2 transcrip-tion[76]. AA can also be converted to leukotrienes (LTs) by the action of 5-LOX (Figure 3). The first step in the 5-LOX cascade consists of activation of the enzyme by 5-LOX-activating protein, which leads to the formation of the LTs and HPETEs. These LTs induce the synthesis and release of other pro-inflammatory mediators such as IL-8 and platelet-activating factor. Several dietary components including curcumin[77], genistein[78], green tea catechins[79], and resver-atrol[80] have been shown to suppress COX-2 and LOXs. Plummer et al. have conducted a dose-escalation pilot study of a standardized formulation of curcuma extract in 15 patients with advanced colorectal cancer, analysis of basal and LPS-induced PGE2 production during treatment demonstrated a trend toward dose-dependent inhibition[81]. Also, Garcea et al have reported that administration of curcumin (3 600 mg) significantly decreased M1G levels in human malignant colorectal tissue but COX-2 protein levels in malignant colorectal tissue were not affected by curcumin[82]. Nitric oxide synthase (NOS) is mainly localized in astrocytes and microglia, and catalyzes the oxidative deamination of L-arginine to produce nitric oxide (NO), a potent pro-inflammatory mediator. Excess production of iNOS-mediated NO is involved in inflammatory and immunological disorders, pain, neurological diseases, atherosclerosis, and cancer. Several phytochemicals and dietary agents have been investigated for their effects on NOS. Recently, Lee et al have reported that 7-carboxymethyloxy-3',4',5’-trimethoxyflavone have inhibitory effects on Helicobacter pylori-induced iNOS expression and NF-κB activation in AGS human gastric cancer cells[83]. Also, Kim et al found that Japanese plants strongly inhibited LPS- and IFN-γ-stimulated NO generation in RAW 264.7 murine macrophages[84].
Animal models in carcinogenesis and cancer chemoprevention
Given the fact that cancer development can be roughly divided into three stages of initiation, promotion and progression, control over this disease may only be achieved if molecular changes underlying each stage can be well-characterized and recapitulated in animal models of carcino-genesis. Hence, animal models are critically important and play a central role in expediting the development of new chemopreventive approaches and therapies for cancer.
Genetically engineered mouse models of cancer Once the efficacy of the chemopreventive agents has been established in nude mice the next logical step would be to attain to good understanding of the molecular mechanisms underlying such effects and this is where autochthnous, germ-line transgenic and knockout animals come into use. These models are in sharp contrast to in vitro culture or orthotopic transplantation of cell lines or tumors from clinical specimens, as described above, because these autochthnous or trans-genic animals closely mimic the complex interactions that occur within the tumor microenvironment. By virtue of their design, these animals could potentially provide a unique window of opportunity to investigate the molecular events related to the various stages of cancer development. In other words, these animal models could potentially recapitulate some of the salient features of cancer progression in humans. TRAMP is one such genetically modified mouse model prostatic intraepithelial neoplasia (PIN) that is commonly used to study prostate cancer chemoprevention and has received considerable attention over several past years[85,86]. The TRAMP model was generated with a probasin (PB)-Tag transgene containing the SV-40 early genes (T and t antigens, Tag) under the control of the minimal rat PB gene promoter. This strategy was designed to target the expression of a large T antigen to abrogate the function of p53 and Rb tumor suppressor genes and to target small t antigens to inactivate protein phosphatase 2A[87]. The TRAMP transgenic mice develop high-grade PIN within 8–12 weeks of birth and ultimately develop metastases between 23–30 weeks, primarily to the lymph nodes, lungs and livers. That is considered the precursor to invasive carcinoma because it is more often than not associated with the malignant form of the disease and the primary architectural and cytological features of PIN resemble that of invasive carcinoma. Hence, the ability to retard PIN formation is considered as an important merit in agents that are tested against prostate carcinoma[88]. Genis-tein, an abundant soy isoflavone in the diet has been shown to reduce the incidence of poorly differentiated prostatic adenocarcinomas in a dose-dependent manner and down-regulate epidermal growth factor receptor, insulin-like growth factor-1 (IGF-1), and ERK-1 mRNA expressions[89]. Likewise, oral consumption of green tea polyphenols has also been shown to suppress the IGF-1 signaling in TRAMP mice[90]. Cancer phenotypes have also been described in various knockout mice. The Nkx 3.1 homeobox gene has been shown to be essential for prostate differentiation and function. Loss-of-function of Nkx 3.1 results in histopathological defects that resemble prostate cancer initiation and progression in humans. These mutant mice represent yet another excellent model to study prostate cancer initiation.
Familial adenomatous polyposis (FAP) is a typical phenotype that involves the development of several adenomatous polyps carpeting the colonic mucosa. In its classical form, almost 100% of these adenomatous polyps are considered cancer prone. FAP is often the result of mutations or deletions of the adenomatous polyposis coli (Apc) gene. Because rodents almost never develop spontaneous cancer in the colon, they often need to be induced or genetically manipulated to develop colon cancer. This led to the development of Apc (+/–) min mice that almost spontaneously develop pre-neoplastic intestinal polyps. These mice present an excellent model to study intestinal and colon cancer chemoprevention because alterations in the number of polyps can be detected with great ease. Recent work from our laboratory and our unpublished results have shown that low doses of 300 ppm sulforaphane and 0.5% dibenzoylmethane (DBM) incorporated in the diet can significantly inhibit polyp formation in Apc min mice[5]. However adenocarcinomas are seldom observed and the progression from aberrant crypt foci (ACF) to aggressive carcinoma is not well established in this model. Another rodent model that uses carcinogen-dimethylhydrazine to induce colon cancer is the azoxymeth-ane (AOM) rat model. Dimethylhydrazine is metabolized to AOM and AOM-induced tumors share many histopathological characteristics with human tumors. They, like human tumors, are often mutated on K-ras and β-catenin genes and display microsatellite instability, but, unlike human colon cancers, seldom show the mutated Apc gene. This model allows intervention by dietary phytochemicals prior to each stage of cancer development and thus, proves to be yet another valuable tool to study colon cancer chemopre-vention. Both the Apc min and AOM rat models of colon cancers could be integrated into other common progression and promotion models of colon cancer. As an example, Tanaka et al. investigated the effects of dextran sodium sulfate (DSS) treatment in the development of colonic neoplasms in Apc min mice. Apc (+/–) and Apc (+/+) mice were exposed to 2% DSS in drinking water for 7 days and such treatment greatly increased the inflammation scores associated with high levels of β-catenin, cyclooxygenase-2, iNOS and nitrotyrosine. These results suggest that DSS has a strong promotion effect in intestinal carcinogenesis of Apc min mice[92,93]. Most recently, this DSS-induced inflammatory model has been applied to our Nrf2 –/– mice[94] and the results from this study will be discussed later on in this review.
Xenograft models of cancer A very quick though rather non-mechanistic approach to evaluating the efficacy of chemopreventive agents in tumor suppression entails the use of immune suppressed athymic nude mice. Nude mice often represent a routinely used valuable research tool because they can receive many types of tissue and tumor grafts and they offer no rejection response. The effects of the chemopreventive agents on the xenograft can be easily monitored by measuring the dimensions and the volume of the tumor graft. Innumerable researchers have used this model to evaluate the effectiveness of various anti-cancer agents. Research from our laboratory has clearly demonstrated that a combination of dietary agents curcumin and PEITC can effectively retard tumor formation in nude mice bearing human prostate cancer PC-3 xenografts and such inhibition was found at least in part to suppress the Akt and NF-κB signaling pathways, ultimately resulting in enhanced expression of apoptosis biomarkers caspase-3 and caspase-9[14]. Apigenin, a naturally occurring flavonoid compound has also been shown to suppress hypoxia-inducible factor-1 (HIF-1) and vascular endothelial growth factor (VEGF) expression in tumor tissues of nude mice bearing A549 human lung cancer cell xenografts[95]. There are many other examples of using nude mice xenografts in cancer chemopre-vention research and these could be found in some of the companion articles in this Special Issue such as the articles by Drs Agarwal and Sarkar[96,97].
Knockout rodent models of cancer To date, most of the knockout rodent models are tissue- or site-specific. However, it is well understood that most cancers are multi-factorial and arise from multiple mutations. Hence, knocking out a gene that exerts pleitropic effects or is central to the development of several cancers presents a model that offers a more mechanistic approach to cancer development and chemoprevention. Nuclear factor E2 related factor 2, Nrf2, as described above is a master transcription factor that has been shown to regulate the expression of more than 200 genes, including those involved in Phase II detoxifying and antioxidant genes[20,98,99]. One of the most prevalent mechanisms by which dietary phytochemicals effectively halt cancer initiation, formation and development is by manipulating the levels of detoxifying and antioxidant cellular defense enzymes. This manipulation mainly involves the regulation via Nrf2 of the ARE that flanks the 5’-flanking region of almost all detoxifying and antioxidant response elements. Hence the use of Nrf2 disrupted mice sheds light on the mechanistic details of cancer chemoprevention by these dietary agents. Because Nrf2 exerts pleiotropic effects, mice that lack this gene are spontaneously predisposed to a variety of carcinogen-induced cancers. Subjecting these mice to the putative chemopreventive agents aids in elucidating the role of Nrf2 in chemoprevention. SFN and dithiolthiones have been shown to transcriptionally induce the expression of detoxifying and antioxidant enzymes through the activation of Nrf2[100]. SFN blocked carcinogen benzo[a]pyrene-induced forestomach tumors and this effect was abrogated in mice lacking the Nrf2 gene[101]. The Nrf2 knockout mouse model allows integration with other cancer models. Recent work from our laboratory established that Nrf2-deficient mice are more susceptible to DSS-induced colitis and this was associated with increased levels of pro-inflammatory mediators such as COX-2, iNOS, IL-6 and TNF-α and a concomitant decrease in antioxidant enzymes such as HO-1, NAD(P)H quinone oxidoreductase 1 (NQO1), UGT and GST[94]. The Nrf2 knockout mouse model has been successfully integrated into skin cancer models that involve initiation with DMBA and promotion by TPA. Work from our laboratory has demonstrated that Nrf2-deficient mice are greatly susceptible to the development of skin cancer by DMBA/TPA applications as compared to the wild type mice and importantly dietary agent SFN, which could effectively suppress carcinogenesis in the wild type mice in part by enhancing the expression of the antioxidant enzyme HO-1, lost its preventive effect in the Nrf2 –/– mice[102]. Surprisingly, in the skin tumors of DMBA/TPA treated Nrf2+/+ mice, there was a loss of expression of both Nrf2 and HO-1 proteins (Yu et al,unpublished data), suggesting potential methylation of the CpG island of Nrf2, thereby inactivating Nrf2 expression with the subsequent decrease in expression of defensive enzymes such as HO-1, that may contribute to the overall carcinogenesis[102].
Each animal model discussed above has immense relevance and potential application in the field of cancer chemoprevention and these animal models will undoubtedly facilitate our ability to discover other dietary factors or synthetic compounds that would be effective at preventing cancer as well as the specific stage(s) of carcinogenesis at which they become effective. Almost every model discussed may seem imperfect in one sense or another, however, each model has contributed significantly to our current knowledge of chemoprevention. Furthermore, cross-breeding to obtain double or triple knockout mice may also greatly help in elucidating the mechanism of chemoprevention by dietary agents in the future. The primary objective for the use of animal models is to ultimately understand the causal relationship between human exposure to dietary phytochemicals and reduced cancer risk. Although many studies have shown the protective effects of these phytochemicals in human clinical trials, not many of these agents have received FDA approval. Some of the main challenges are that while the beneficial effects of these agents are very well studied, the possible adverse side-effects of these compounds are lagging behind, although their elucidation is being pursued at a rapid pace. These adverse side-effects may in part be due to the ability of these agents to cause various drug to drug interactions due to unwarranted inhibition or activation of various drug metabolizing enzymes other than the detoxifying enzymes. Successful prediction of the consequences of human consumption of these phytochemicals will be possible only after appropriate doses are designed and the safety issues are taken care of.
Conclusion
The modulation of cellular signal transduction pathways by naturally occurring phytochemicals has recently been extended to elucidate the molecular basis of cancer chemopre-vention with dietary factors. Dietary chemopreventive agents derived from the human dietary fruits, vegetables and tea and coffee beverages have gained much attention recently, in lieu of the fact that cancer incidence remains high throughout the world as well as in China (http://english.people.com.cn/200705/21/eng20070521_376395.html) and the concept of cancer intervention by means of dietary phytochemicals needs to be more widely accepted. Because of the rapid progress in our understanding of the genetic and epigenetic mechanisms of carcinogenesis at the signal transduction level, additional molecular targets have been and will be identified for the potential intervention at the initiation, promotion and progression steps of carcinogenesis and cancer development. So far, these include transcription factors such as Nrf2, NF-κB, AP-1, and MAPKs. The signaling pathways mediated by these transcription factors in response to pro-inflammatory cytokines, growth factors, xenobiotics, oxidative stress and tumor promoters can regulate a wide array of genes involved in many cellular events such as cell cycle control, differentiation, transformation, apoptosis and tumorigenesis. Because of their potential role in preventing cancer development, the mechanisms by which phytochemicals regulate signaling pathways have been extensively studied. Dietary chemopreventive agents activate Nrf2-mediated gene expression either by directly modifying the cysteine residues on Keap1 to disrupt the Nrf2-Keap1 complex or by activating kinase signaling pathways such as MAPKs, protein kinase C (PKC), and PI3K to phosphorylate Nrf2/Keap1 complex and/or facilitate the release of Nrf2 or to increase the nuclear translocation of Nrf2 and regulate the transcriptional activity of Nrf2 nuclear co-activators. While many chemopreventive agents directly act on the Nrf2/ARE signaling pathway to induce detoxification and antioxidant enzyme systems, which may block tumor initiation in normal cells, the same compounds and/or others may exert anti-promotion or anti-progression effects by modulating the NF-κB and AP-1 signaling pathways in abnormal pre-initiated cancer cells[103]. Cellular signaling cascades mediated by NF-κB and AP-1 generally act as key regulators of many of the aforementioned biochemical processes. Results from both in vitro and in vivo studies suggest that phytochemicals can inhibit the activation of NF-κB and/or AP-1 in a cell- or tissue-specific manner. Although experimental data from cell culture models provide valuable information regarding the molecular and cellular mechanisms involved in the modulation of Nrf2, NF-κB and AP-1 signaling pathways, the precise chemical mechanisms and the signal transduction cascades between the interplay of phytochemicals and transcription factors are still not fully understood. For instance, although NF-κB and AP-1 may be considered as prime molecular targets for chemoprevention by dietary phytochemicals, cell type- and stimuli-dependency between these two transcription factors often makes it complicated to deduce a unique mechanism for the chemopreventive activity of phyto-chemicals. While chemoprevention studies using animal models certainly provide promising results for the chemopreventive agents discussed in this review article, future confirmatory human clinical trials coupled with epidemiological data would be needed to support their eventual chemopreventive potentials. The modulation of the above signaling pathways, transcription factors and genes expression by cancer chemopreventive phytochemicals would provide potential opportunities for future design of chemopreventive agents based on molecular targeting.
Acknowledgments
The authors are grateful to all members of Dr Ah-Ng Tony Kong’s laboratory as well as researchers in the scientific community whose works have been cited here. The authors also regret the inability to include many other excellent relevant citations in the interests of comprehensive abridgement.
References
- Sporn MB, Dunlop NM, Newton DL, Smith JM. Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids). Fed Proc 1976;35:1332-8.
- Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer 2003;3:768-80.
- Chen C, Kong AN. Dietary cancer-chemopreventive compounds: from signaling and gene expression to pharmacological effects. Trends Pharmacol Sci 2005;26:318-26.
- Hu R, Kong AN. Activation of MAP kinases, apoptosis and nutrigenomics of gene expression elicited by dietary cancer-prevention compounds. Nutrition 2004;20:83-8.
- Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C, et al. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis 2006;27:2038-46.
- Jeong WS, Keum YS, Chen C, Jain MR, Shen G, Kim JH, et al. Differential expression and stability of endogenous nuclear factor E2-related factor 2 (Nrf2) by natural chemopreventive compounds in HepG2 human hepatoma cells. J Biochem Mol Biol 2005;38:167-76.
- Khor TO, Hu R, Shen G, Jeong WS, Hebbar V, Chen C, et al. Pharmacogenomics of cancer chemopreventive isothiocyanate compound sulforaphane in the intestinal polyps of ApcMin/+ mice. Biopharm Drug Dispos 2006;27:407-20.
- Singletary K, MacDonald C. Inhibition of benzo[a]pyrene- and 1,6-dinitropyrene-DNA adduct formation in human mammary epithelial cells bydibenzoylmethane and sulforaphane. Cancer Lett 2000;155:47-54.
- Huang MT, Wang ZY, Georgiadis CA, Laskin JD, Conney AH. Inhibitory effects of curcumin on tumor initiation by benzo[a]pyrene and 7,12-dimethylbenz[a]anthracene. Carcinogenesis 1992;13:2183-6.
- Hu R, Kim BR, Chen C, Hebbar V, Kong AN. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis 2003;24:1361-7.
- Yu R, Mandlekar S, Harvey KJ, Ucker DS, Kong AN. Chemo-preventive isothiocyanates induce apoptosis and caspase-3-like protease activity. Cancer Res 1998;58:402-8.
- Xu C, Shen G, Chen C, Gelinas C, Kong AN. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene 2005;24:4486-95.
- Kim JH, Xu C, Keum YS, Reddy B, Conney A, Kong AN. Inhibition of EGFR signaling in human prostate cancer PC-3 cells by combination treatment with beta-phenylethyl isothiocyanate and curcumin. Carcinogenesis 2006;27:475-82.
- Khor TO, Keum YS, Lin W, Kim JH, Hu R, Shen G, et al. Combined inhibitory effects of curcumin and phenethyl isothiocyanate on the growth of human PC-3 prostate xenografts in immunodeficient mice. Cancer Res 2006;66:613-21.
- Chen C, Shen G, Hebbar V, Hu R, Owuor ED, Kong AN. Epigallocatechin-3-gallate-induced stress signals in HT-29 human colon adenocarcinoma cells. Carcinogenesis 2003;24:1369-78.
- Qin J, Xie LP, Zheng XY, Wang YB, Bai Y, Shen HF, et al. A component of green tea, (-)-epigallocatechin-3-gallate, promotes apoptosis in T24 human bladder cancer cells via modulation of the PI3K/Akt pathway and Bcl-2 family proteins. Biochem Biophys Res Commun 2007;354:852-7.
- Keum YS, Yu S, Chang PP, Yuan X, Kim JH, Xu C, et al. Mechanism of action of sulforaphane: inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res 2006;66:8804-13.
- Maheo K, Morel F, Langouet S, Kramer H, Le Ferrec E, Ketterer B, et al. Inhibition of cytochromes P-450 and induction of glutathione S-transferases by sulforaphane in primary human and rat hepatocytes. Cancer Res 1997;57:3649-52.
- Shen G, Xu C, Hu R, Jain MR, Gopalkrishnan A, Nair S, et al. Modulation of nuclear factor E2-related factor 2-mediated gene expression in mice liver and small intestine by cancer chemo-preventive agent curcumin. Mol Cancer Ther 2006;5:39-51.
- Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, et al. Identification of Nrf2-regulated genes induced by chemopreven-tive isothiocyanate PEITC by oligonucleotide microarray. Life Sci 2006;79:1944-55.
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem 2000;275:16023-9.
- Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharma-col 2001;173:154-60.
- Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O’Connor T, et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci 2001;59:169-77.
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci USA 2001;98:3410-5.
- Hayes JD, Chanas SA, Henderson CJ, McMahon M, Sun C, Moffat GJ, et al. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem Soc Trans 2000;28:33-41.
- Morimitsu Y, Nakagawa Y, Hayashi K, Fujii H, Kumagai T, Nakamura Y, et al. A sulforaphane analogue that potently activates the Nrf2-dependent detoxification pathway. J Biol Chem 2002;277:3456-63.
- Balogun E, Hoque M, Gong P, Killeen E, Green CJ, Foresti R, et al. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem J 2003;371:887-95.
- Li W, Yu SW, Kong AN. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J Biol Chem 2006;281:27,251-63.
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999;13:76-86.
- Kobayashi M, Itoh K, Suzuki T, Osanai H, Nishikawa K, Katoh Y, et al. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells 2002;7:807-20.
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 2002;62:5196-203.
- Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci USA 1993;90:2965-9.
- Huang TT, Kudo N, Yoshida M, Miyamoto S. A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/Ikappa-Balpha complexes. Proc Natl Acad Sci USA 2000;97:1014-19.
- Johnson C, Van Antwerp D, Hope TJ. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J 1999;18:6682-93.
- Baichwal VR, Baeuerle PA. Activate NF-kappa B or die? Curr Biol 1997;7:R94-6.
- Bours V, Bentires-Alj M, Hellin AC, Viatour P, Robe P, Delhalle S, et al. Nuclear factor-kappa B, cancer, and apoptosis. Biochem Pharmacol 2000;60:1085-9.
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-kappaB activity. Annu Rev Immunol 2000;18:621-63.
- Hatta Y, Arima N, Machino T, Itoh T, Hashimoto S, Takeuchi J, et al. Mutational analysis of IkappaBalpha in hematologic malignancies. Int J Mol Med 2003;11:239-42.
- Huang S, Pettaway CA, Uehara H, Bucana CD, Fidler IJ. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 2001;20:4188-97.
- Kim DW, Sovak MA, Zanieski G, Nonet G, Romieu-Mourez R, Lau AW, et al. Activation of NF-kappaB/Rel occurs early during neoplastic transformation of mammary cells. Carcinogenesis 2000;21:871-9.
- Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res 1999;5:119-27.
- Bharti AC, Aggarwal BB. Nuclear factor-kappa B and cancer: its role in prevention and therapy. Biochem Pharmacol 2002;64:883-8.
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta 1991;1072:129-57.
- Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 2003;3:859-68.
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol 2002;4:E131-6.
- Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene 2001;20:2390-400.
- Wang ZQ, Grigoriadis AE, Mohle-Steinlein U, Wagner EF. A novel target cell for c-fos-induced oncogenesis: development of chondrogenic tumours in embryonic stem cell chimeras. EMBO J 1991;10:2437-50.
- Eferl R, Ricci R, Kenner L, Zenz R, David JP, Rath M, et al. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell 2003;112:181-92.
- Young MR, Li JJ, Rincon M, Flavell RA, Sathyanarayana BK, Hunziker R, et al. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc Natl Acad Sci USA 1999;96:9827-32.
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001;410:37-40.
- Funakoshi-Tago M, Tago K, Sonoda Y, Tominaga S, Kasahara T. TRAF6 and C-SRC induce synergistic AP-1 activation via PI3-kinase-AKT-JNK pathway. Eur J Biochem 2003;270:1257-68.
- Jeong WS, Kim IW, Hu R, Kong AN. Modulatory properties of various natural chemopreventive agents on the activation of NF-kappaB signaling pathway. Pharm Res 2004;21:661-70.
- Zhu M, Zhang Y, Cooper S, Sikorski E, Rohwer J, Bowden GT. Phase II enzyme inducer, sulforaphane, inhibits UVB-induced AP-1 activation in human keratinocytes by a novel mechanism. Mol Carcinog 2004;41:179-86.
- Jeong WS, Kim IW, Hu R, Kong AN. Modulation of AP-1 by natural chemopreventive compounds in human colon HT-29 cancer cell line. Pharm Res 2004;21:649-60.
- Gonzalez FA, Raden DL, Davis RJ. Identification of substrate recognition determinants for human ERK1 and ERK2 protein kinases. J Biol Chem 1991;266:22159-63.
- Alvarez E, Northwood IC, Gonzalez FA, Latour DA, Seth A, Abate C, et al. Pro-Leu-Ser/Thr-Pro is a consensus primary sequence for substrate protein phosphorylation. Characterization of the phosphorylation of c-myc and c-jun proteins by an epidermal growth factor receptor threonine 669 protein kinase. J Biol Chem 1991;266:15277-85.
- Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem 1995;270:14843-6.
- Cano E, Mahadevan LC. Parallel signal processing among mammalian MAPKs. Trends Biochem Sci 1995;20:117-22.
- Kyriakis JM, Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays 1996;18:567-77.
- Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem 1996;271:31929-36.
- Osborn MT, Chambers TC. Role of the stress-activated/c-Jun NH2-terminal protein kinase pathway in the cellular response to adriamycin and other chemotherapeutic drugs. J Biol Chem 1996;271:30950-5.
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem 1995;270:16483-6.
- Lee KW, Kim JH, Lee HJ, Surh YJ. Curcumin inhibits phorbol ester-induced up-regulation of cyclooxygenase-2 and matrix metalloproteinase-9 by blocking ERK1/2 phosphorylation and NF-kappaB transcriptional activity in MCF10A human breast epithelial cells. Antioxid Redox Signal 2005;7:1612-20.
- Chen YR, Tan TH. Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene 1998;17:173-8.
- Salh B, Assi K, Templeman V, Parhar K, Owen D, Gomez-Munoz A, et al. Curcumin attenuates DNB-induced murine colitis. Am J Physiol Gastrointest Liver Physiol 2003;285:G235-43.
- Li Y, Li X, Sarkar FH. Gene expression profiles of I3C- and DIM-treated PC3 human prostate cancer cells determined by cDNA microarray analysis. J Nutr 2003;133:1011-9.
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 2003;3:745-56.
- Sugarman BJ, Aggarwal BB, Hass PE, Figari IS, Palladino MA Jr, Shepard HM. Recombinant human tumor necrosis factor-alpha: effects on proliferation of normal and transformed cells in vitro. Science 1985;230:943-5.
- Giri DK, Aggarwal BB. Constitutive activation of NF-kappaB causes resistance to apoptosis in human cutaneous T cell lymphoma HuT-78 cells. Autocrine role of tumor necrosis factor and reactive oxygen intermediates. J Biol Chem 1998;273:14008-14.
- Tucker SJ, Rae C, Littlejohn AF, Paul A, MacEwan DJ. Switching leukemia cell phenotype between life and death. Proc Natl Acad Sci USA 2004;101:12940-5.
- Estrov Z, Kurzrock R, Pocsik E, Pathak S, Kantarjian HM, Zipf TF, et al. Lymphotoxin is an autocrine growth factor for Epstein-Barr virus-infected B cell lines. J Exp Med 1993;177:763-74.
- Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med 1999;5:828-31.
- Shishodia S, Amin HM, Lai R, Aggarwal BB. Curcumin (diferuloyl-methane) inhibits constitutive NF-kappaB activation, induces G1/S arrest, suppresses proliferation, and induces apoptosis in mantle cell lymphoma. Biochem Pharmacol 2005;70:700-13.
- Yang F, de Villiers WJ, McClain CJ, Varilek GW. Green tea polyphenols block endotoxin-induced tumor necrosis factor-production and lethality in a murine model. J Nutr 1998;128:2334-40.
- Martin AR, Villegas I, Sanchez-Hidalgo M, de la Lastra CA. The effects of resveratrol, a phytoalexin derived from red wines, on chronic inflammation induced in an experimentally induced colitis model. Br J Pharmacol 2006;147:873-85.
- Subbaramaiah K, Dannenberg AJ. Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends Pharmacol Sci 2003;24:96-102.
- Plummer SM, Holloway KA, Manson MM, Munks RJ, Kaptein A, Farrow S, et al. Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-kappaB activation via the NIK/IKK signalling complex. Oncogene 1999;18:6013-20.
- Mutoh M, Takahashi M, Fukuda K, Matsushima-Hibiya Y, Mutoh H, Sugimura T, et al. Suppression of cyclooxygenase-2 promoter-dependent transcriptional activity in colon cancer cells by chemopreventive agents with a resorcin-type structure. Carcinogenesis 2000;21:959-63.
- Gerhauser C, Klimo K, Heiss E, Neumann I, Gamal-Eldeen A, Knauft J, et al. Mechanism-based in vitro screening of potential cancer chemopreventive agents. Mutat Res 2003;523–4:163-72.
- Subbaramaiah K, Chung WJ, Michaluart P, Telang N, Tanabe T, Inoue H, et al. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J Biol Chem 1998;273:21875-82.
- Plummer SM, Hill KA, Festing MF, Steward WP, Gescher AJ, Sharma RA. Clinical development of leukocyte cyclooxygenase 2 activity as a systemic biomarker for cancer chemopreventive agents. Cancer Epidemiol Biomarkers Prev 2001;10:1295-9.
- Garcea G, Berry DP, Jones DJ, Singh R, Dennison AR, Farmer PB, et al. Consumption of the putative chemopreventive agent curcumin by cancer patients: assessment of curcumin levels in the colorectum and their pharmacodynamic consequences. Cancer Epidemiol Biomarkers Prev 2005;14:120-5.
- Lee JS, Kim HS, Hahm KB, Sohn MW, Yoo M, Johnson JA, et al. Inhibitory effects of 7-carboxymethyloxy-3',4',5-trimethoxy-flavone (DA-6034) on Helicobacter pylori-induced NF-kappa B activation and iNOS expression in AGS cells. Ann N Y Acad Sci 2007;1095:527-35.
- Kim OK, Murakami A, Nakamura Y, Ohigashi H. Screening of edible Japanese plants for nitric oxide generation inhibitory activities in RAW 264.7 cells. Cancer Lett 1998;125:199-207.
- Greenberg NM, DeMayo FJ, Sheppard PC, Barrios R, Lebovitz R, Finegold M, et al. The rat probasin gene promoter directs hormonally and developmentally regulated expression of a heterologous gene specifically to the prostate in transgenic mice. Mol Endocrinol 1994;8:230-9.
- Klein RD. The use of genetically engineered mouse models of prostate cancer for nutrition and cancer chemoprevention research. Mutat Res 2005;576:111-9.
- Hernandez I, Maddison LA, Wei Y, DeMayo F, Petras T, Li B, et al. Prostate-specific expression of p53(R172L) differentially regulates p21, Bax, and mdm2 to inhibit prostate cancer progres-sion and prolong survival. Mol Cancer Res 2003;1:1036-47.
- Kaplan-Lefko PJ, Chen TM, Ittmann MM, Barrios RJ, Ayala GE, Huss WJ, et al. Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. Prostate 2003;55:219-37.
- Mentor-Marcel R, Lamartiniere CA, Eltoum IE, Greenberg NM, Elgavish A. Genistein in the diet reduces the incidence of poorly differentiated prostatic adenocarcinoma in transgenic mice (TRAMP). Cancer Res 2001;61:6777-82.
- Adhami VM, Siddiqui IA, Ahmad N, Gupta S, Mukhtar H. Oral consumption of green tea polyphenols inhibits insulin-like growth factor-I-induced signaling in an autochthonous mouse model of prostate cancer. Cancer Res 2004;64:8715-22.
- Bieberich CJ, Fujita K, He WW, Jay G. Prostate-specific and androgen-dependent expression of a novel homeobox gene. J Biol Chem 1996;271:31779-82.
- Tanaka T, Kohno H, Suzuki R, Hata K, Sugie S, Niho N, et al. Dextran sodium sulfate strongly promotes colorectal carcinogenesis in Apc(Min/+) mice: inflammatory stimuli by dextran sodium sulfate results in development of multiple colonic neoplasms. Int J Cancer 2006;118:25-34.
- Hirono I, Ueno I, Aiso S, Yamaji T, Golberg L. Enhancing effect of dextran sulfate sodium on colorectal carcinogenesis by 1,2-dimethylhydrazine in rats. Gann 1983;74:493-6.
- Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res 2006;66:11580-4.
- Liu LZ, Fang J, Zhou Q, Hu X, Shi X, Jiang BH. Apigenin inhibits expression of vascular endothelial growth factor and angiogenesis in human lung cancer cells: implication of chemo-prevention of lung cancer. Mol Pharmacol 2005;68:635-43.
- Singh RP, Agarwal R. Mechanisms of action of novel agents for prostate cancer chemoprevention. Endocr Relat Cancer 2006;13:751-78.
- Sarkar FH, Li Y. The role of isoflavones in cancer chemopre-vention. Front Biosci 2004;9:2714-24.
- Nair S, Xu C, Shen G, Hebbar V, Gopalakrishnan A, Hu R, et al. Toxicogenomics of endoplasmic reticulum stress inducer tunicamycin in the small intestine and liver of Nrf2 knockout and C57BL/6J mice. Toxicol Lett 2007;168:21-39.
- Osburn WO, Wakabayashi N, Misra V, Nilles T, Biswal S, Trush MA, et al. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch Biochem Biophys 2006;454:7-15.
- Yu X, Kensler T. Nrf2 as a target for cancer chemoprevention. Mutat Res 2005;591:93-102.
- Fahey JW, Haristoy X, Dolan PM, Kensler TW, Scholtus I, Stephenson KK, et al. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci USA 2002;99:7610-5.
- Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res 2006;66:8293-6.
- Nair S, Li W, Kong AN. Natural dietary anti-cancer chemo-preventive compounds: redox-mediated differential signaling mechanisms in cytoprotection of normal cells versus cytotoxicity in tumor cells. Acta Pharmacol Sin 2007;28:459-72.