
Keap1 eye on the target: chemoprevention of liver cancer1
Introduction
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide, causing nearly 600 000 deaths each year. More than half of these deaths occur in China. Residents of Qidong, Jiangsu Province, China are at particularly high risk. In this area, HCC is the leading cause of cancer death. Exposure to dietary aflatoxins and chronic infection with hepatitis B virus (HBV) may contribute to this extraordinarily increased risk[1]. Aflatoxins are mycotoxins produced by the fungus Aspergillus flavus, which are found in contaminated foods such as corn and nuts. Aflatoxin exposure and infection with HBV may be responsible for 90% or more of the HCC cases in this area[2]. Prevention strategies targeting HBV and aflatoxin exposure could drastically impact rates of HCC. For example, since a universal HBV vaccination program was implemented in Taiwan in 1984, the incidence of HCC in children has declined[3]. Aflatoxin exposure can be reduced by improving methods used to grow, harvest, store, and process susceptible foods. This could include the control of the moisture content and relative humidity for food storage, sorting strategies to remove contaminated items, or dietary changes, such as shifting to foods which are less susceptible to aflatoxin contamination (for example rice instead of corn)[4]. However, the complete elimination of aflatoxin contamination might not be possible. For this reason, chemoprevention is a practical strategy to reduce the incidence of HCC in populations with high dietary aflatoxin exposure.
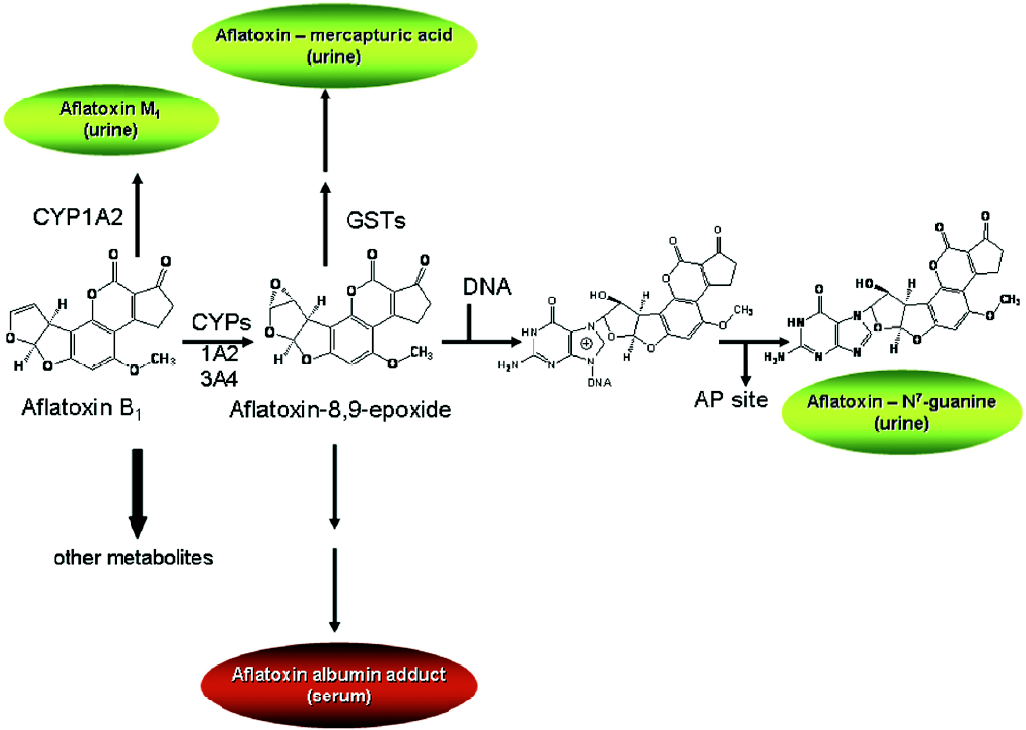
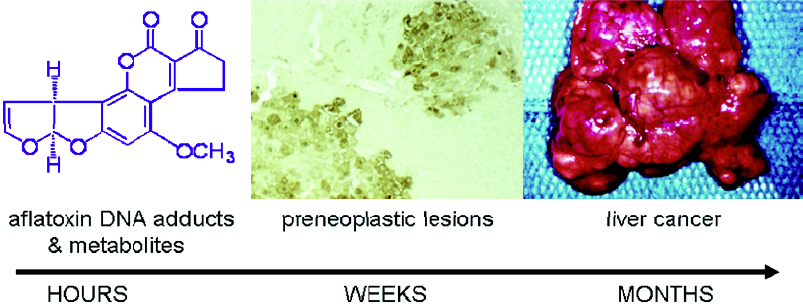
Aflatoxin metabolism and mechanisms of aflatoxin-induced hepatocarcinogenesis are well known (Figure 1). This knowledge provides the basis for evaluation of exposure to aflatoxin, as well as modulation of aflatoxin disposition by chemopreventive agents. Aflatoxin B1 is metabolized by cytochrome P450s[5,6] to a reactive epoxide (aflatoxin-8,9-epoxide) and other oxidation products, including aflatoxin M1. The epoxide can react further by interacting with DNA to form a promutagenic aflatoxin-N7-guanine adduct. This is an unstable adduct which rapidly undergoes depurination and excretion in urine[7]. The epoxide can also react with serum albumin to form long-lived lysine adducts[8]. In addition, the epoxide can be conjugated by glutathione S-transferases (GSTs), which are further metabolized to form aflatoxin-mercapturic acid detoxification products that can be excreted in urine[9]. Products of aflatoxin DNA damage and toxicity as well as other metabolites can be used as biomarkers to evaluate the modulation of aflatoxin activation and detoxication. For example, levels of both lysine and N7-guanine adducts, which are excreted in urine, have been shown to correlate with aflatoxin exposure[10–13]. In animals, changes in hepatic aflatoxin-N7-guanine adduct formation are associated with the degree of chemoprotection as measured by reduction in preneoplastic foci and cancers (Figure 2)[14,15]. In addition, the levels of aflatoxin M1 excreted in urine have been shown to reflect human exposure[16] and predict chemopreventive efficacy and liver cancer risk[17–19]. Furthermore, increased formation of aflatoxin-mercapturic acid metabolites can be measured in the urine of laboratory animals as well as humans and is inversely associated with levels of aflatoxin DNA adducts formed in liver and excreted into urine. These biomarkers are critical tools for evaluation of chemopreven-tive agents in animal models and clinical interventions.
Mechanisms for prevention
Induction of cytoprotective enzymes Induction of xeno-biotic metabolism enzymes has been shown to be an effective means of protection against carcinogenesis, mutagenesis, and other toxicities mediated by electrophiles. These cyto-protective enzymes (often referred to as “phase 2 enzymes”) include conjugating and antioxidative enzymes. Conjugating enzymes, such as GSTs and UDP-glucuronosyl transferases facilitate the elimination of carcinogens. Additional protection is achieved through induction of antioxidative enzymes that enhance the resistance of cells to oxidative stress. Induction of phase 2 enzymes has been shown to be sufficient for chemoprevention. Known chemopreventive agents, such as oltipraz and sulforaphane, enhance the expression of many of these detoxication and cytoprotective genes.
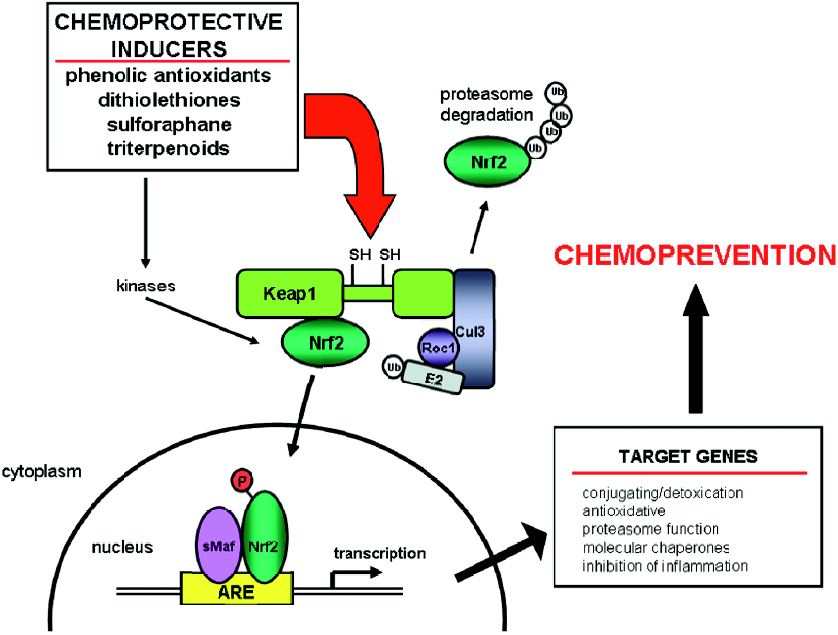
Nrf2 activation Nrf2 (NF–E2-related factor 2) is a member of the basic leucine zipper NF–E2 family of transcription factors. Studies using Nrf2-deficient mice have defined the crucial role of Nrf2 in chemoprevention (Figure 3). Nrf2-deficient mice are more susceptible to toxicity, DNA adduct formation, and cancer development in several models of chemical-induced carcinogenesis. While the basal expression of some cytoprotective genes is Nrf2 dependent[20–24], the increased sensitivity caused by loss of Nrf2 is likely due to an impaired ability to mount an adaptive response in the face of repetitive carcinogenic challenges through induction of a broad array of cytoprotective genes[25–28]. For example, DNA adduct formation is increased in Nrf2-deficient mice compared to wild-type mice following exposure to carcinogens, such as diesel exhaust[29], aflatoxin B1[30], and benzo[a]pyrene[31]. Nrf2-deficient mice develop a higher burden of gastric neoplasia following treatment with benzo[a]pyrene compared to wild-type mice[20]. Compared to wild-type mice, Nrf2-null mice also have increased incidence of skin tumors and tumor numbers per mouse in a 7,12-dime-thylbenz(a)anthracene-induced skin tumorigenesis model[32]. Chemopreventive agents, such as oltipraz and sulforaphane, do not induce cytoprotective genes in Nrf2-deficient mice. Moreover, the antitumorigenic actions of these agents are lost in Nrf2-disrupted mice[20,32]. Nrf2 regulates a larger cytoprotective response beyond the conjugating and anti-oxidative classes. This response includes the ubiquitin/proteasome system, the molecular chaperones/stress response system, and anti-inflammatory responses[27]. This broad protective response makes Nrf2 and its interacting partners important targets for chemoprevention.
Antioxidant response element-mediated regulation of Nrf2 The 5'-flanking region of many cytoprotective and detoxifying enzymes contain a common regulatory element, the antioxidant response element (ARE). Many cytopro-tective genes, such as rat and mouse GST, rat and human NAD(P)H:quinone oxidoreductase (NQO1), and human glutamate cysteine ligase subunits contain AREs[33–37]. Activation of these AREs is induced by a structurally diverse group of chemicals, including oxidizable diphenols, dithiolethiones, isothiocyanates, and Michael acceptors (olefins or acetylenes conjugated to electron-withdrawing groups)[38,39]. This activation occurs through Kelch ECH-associating protein 1 (Keap1)–Nrf2–ARE signaling (Figure 3). Keap1 is an actin binding protein which interacts with the N-terminal Neh2 domain of Nrf2. Keap1 sequesters Nrf2 in the cytoplasm, causing transcriptional repression by preventing the nuclear translocation of Nrf2[40]. Under homeostatic conditions, Keap1 enhances the rate of the proteasomal degradation of Nrf2. Keap1 also acts as an adaptor for the Cullin 3-based E3 ubiquitin ligase[41–43], which acts as a scaffold to form an E3 ligase complex and recruit the ubiquitin-conjugation (E2) enzymes. Oxidative stress may disrupt the Keap1–Nrf2 interaction resulting in the stabilization of Nrf2 and the accumulation of Nrf2 within the cell[44]. Keap1 contains reactive cysteine residues which may act as sensors for electrophilic and oxidative stresses, as well as the chemical inducers described earlier[45]. Cysteine modification may induce a conformational change in Keap1 causing the dissociation of Nrf2. Nrf2 can then translocate to the nucleus where it heterodimerizes with small Maf proteins and binds to the ARE to act as a transcriptional activator[22]. Recent studies have suggested an alternative mechanism where nuclear accumulation of Nrf2 caused by electrophilic stress requires de novo protein synthesis[46]. In addition, cysteine residues in Keap1, which do not participate in the Keap1–Nrf2 interaction, are critical to the degradation of Nrf2 by the ubiquitin–proteasome system[46]. These studies suggest that activation of Nrf2 occurs by impairing the Keap1-mediated degradation of Nrf2.
Other mechanisms of Nrf2 regulation (kinases) Other signaling pathways influence Keap1–Nrf2–ARE signaling through post-transcriptional modification. Several kinase pathways, including protein kinase C (PKC), mitogen-activated protein kinase, phosphatidylinositol 3-kinase (PI3K), and PKR-like endoplasmic reticulum kinase (PERK) have been shown to influence Keap1–Nrf2–ARE signaling[47–49]. For example, phosphorylation of Nrf2 by PKC promotes release from Keap1[50]. Inhibition of PI3K attenuates the nuclear translocation of Nrf2 and transcription of ARE-regulated genes in vitro[51]. PERK phosphorylates Nrf2 and triggers dissociation from Keap1 resulting in increased nuclear translocation[52]. These in vitro studies require further study to determine the significance of these pathways in vivo and their suitability as pharmacological targets for modulating Nrf2 signaling.
Protection in animal models
Several classes of chemical inducers of Nrf2 signaling have been evaluated in animal models of chemical carcinogenesis. These agents include phenolic antioxidants used as food preservatives; dithiolethiones; isothiocyanates, such as sulforaphane; and highly potent synthetic analogs of naturally occurring triterpenoids. These agents induce Nrf2-regulated cytoprotective enzymes and prevent chemically-induced carcinogenesis (Table 1).

Full table
Phenolic antioxidants Phenolic antioxidants, such as butylated hydroxyanisole (BHA) and butylated hydroxytoluene (BHT), are added to foods to prevent oxygen-induced lipid peroxidation. Rats fed diets containing 0.45% BHA or BHT showed increased hepatic GST and UDP-glucuronosyl transferase enzyme activity. In addition, 0.45% BHA or BHT in the diet also reduced aflatoxin-DNA adduct formation in the liver by 85% and 65%, respectively[53]. Dietary administration of BHA or BHT also reduces neoplasia in chemically-induced carcinogenesis models targeting the forestomach, lung, small intestine, breast, and skin[54]. Another antioxidant, ethoxyquin, is also used as a preservative in food. Ethoxyquin given in the diet at 0.05% or 0.5% inhibited liver carcinogenesis in rats exposed to aflatoxin B1[55]. It was subsequently shown that the induction of hepatic cytoprotective enzymes by these antioxidants is mediated by Nrf2 signaling[56,57].
Dithiolethiones Some members of the dithiolethione class of chemopreventive agents are more potent inducers of conjugating and detoxication enzymes than the phenolic antioxidants. The most studied member of this class is 5-(2-pyrazinyl)-4-methyl-1,2-dithiole-3-thione, or oltipraz. During studies evaluating oltipraz for its antischistosomal activity, it was noted that oltipraz caused an increase in glutathione levels in the liver, kidney, and forestomach of mice. Furthermore, dithiolethiones increased the activity of conjugating and detoxication enzymes such as GST, NQO1, and glutathione reductase[58]. It was predicted that oltipraz would be an effective chemopreventive agent due to this induction. Subsequently, oltipraz was shown to be an effective chemopreventive agent in chemical-induced cancer models. Several studies have shown that oltipraz protects against aflatoxin-induced hepatic tumorigenesis. Rats were fed 0.01%–0.1% oltipraz for 4 weeks. On the second and third week of oltipraz feeding, the rats were gavaged with aflatoxin B1 5 times per week. Twelve weeks after oltipraz feeding ended, the rats were killed and liver sections were analyzed for preneoplastic foci. All of the doses of oltipraz reduce the volume of the liver occupied by preneoplastic foci by more than 90%. Hepatic aflatoxin-DNA adduct formation is reduced by 40%–80% over the dose range of 0.01%–0.1%. Furthermore, oltipraz increases hepatic GST enzyme activity, which acts to increase detoxication of the ultimate carcinogenic form of aflatoxin, aflatoxin-8,9-epoxide[59]. These studies were extended to chemopre-vention of HCC induced by aflatoxin in a 2 year study. While aflatoxin-exposed rats receiving the control diet developed HCC or hepatocellular adenomas, dietary oltipraz at 0.075% provided complete protection against both aflatoxin-induced hepatocellular neoplasms. The rats in the oltipraz group also had a significantly longer life span and an increased survival free of liver tumors[60].
The costly synthesis and purification of oltipraz as well as toxicities noted in clinical trials for the chemotherapy of schistosomiasis[61] have indicated that further development of oltipraz could be problematic. A second generation of dithiolethione analogs was evaluated to identify analogs which would be more suitable than oltipraz for further development. The chemopreventive potential of these analogs was evaluated by measuring inhibition of formation of DNA adducts and formation of preneoplastic lesions in the livers of rats treated with aflatoxin B1. 3H-1,2-dithiole-3-thione (D3T) is the most potent with a greater than 90% reduction in DNA adduct formation at the highest dose (0.3 mmol/kg body weight) compared to an approximately 60% reduction by oltipraz at the same dose[62]. Further studies have shown that D3T is also the most potent inhibitor of hepatic preneoplastic lesions[62].
The chemoprotective activity of dithiolethiones is Nrf2-mediated. Increased accumulation of Nrf2 has been shown in extracts from hepatic nuclei of D3T-treated mice[28]. While oltipraz induces the cytoprotective ferritin H and L genes in wild-type mouse embryonic fibroblasts (MEFs), this induction is eliminated in Nrf2-deficient MEFs[63]. In addition, oltipraz and D3T do not induce GSTs and NQO1 in Nrf2-deficient mice[20,28]. A microarray analysis using wild-type and Nrf2-deficient mice showed that D3T induces a large number of Nrf2-dependent genes. In addition to the previously identified Nrf2-dependent detoxication and antioxida-tive genes, new gene clusters were also identified as D3T-inducible and Nrf2-dependent. These include other groups of cytoprotec-tive genes involved in the ubiquitin/protea-some system and molecular chaperones[27]. Furthermore, in chemical-induced cancer models targeting the bladder and forestomach, the chemopreventive efficacy of oltipraz is lost in Nrf2-deficient mice[20,64].
Sulforaphane Glucosinolates are found in high concentrations in cruciferous vegetables. Glucosinolates can be hydrolyzed by myrosinase (an enzyme which is released when the plant is chewed or in the intestinal microflora) to produce isothiocyanates. Isothiocyanates are potent inducers of cytoprotective and detoxication enzymes. Sulforaphane is a potent isothiocyanate whose precursor is abundant in broccoli, particularly in 3 d-old broccoli sprouts. Sulfora-phane induces detoxication and cytoprotective enzymes, such as GSTs and NQO1, in a Nrf2-dependent manner[65–67]. Sulforaphane has been shown to directly interact with Keap1[68]. Furthermore, sulforaphane reduces mammary tumor incidence and multiplicity in rats treated with DMBA[69].
Triterpenoids Synthetic triterpenoids have recently been developed at Dartmouth Medical School (Hanover, NH, USA) as anti-inflammatory agents. These agents are synthetic derivatives of oleanolic acid, a plant-derived compound used in traditional Asian medicine for its weak anti-inflammatory and antitumorigenic activity[70,71]. The synthetic optimization of these triterpenoids has greatly improved upon the weak activity of the naturally occurring compound. These compounds are also being evaluated for both cancer chemotherapy and chemoprevention. In cancer cell lines, these triterpenoids induce differentiation, inhibit growth, induce cell cycle arrest, and induce apoptosis[72–76]. An imidazolide triterpenoid, 1-(2-cyano-3,12-dioxooleana-1,9[11]-dien-28-oyl)imidazole (CDDO-Im), is one of the most potent triterpenoids. While optimizing the synthetic triterpenoids to maximize anti-inflammatory activity[77–79], the presence of Michael acceptor groups was determined to be essential for anti-inflammatory activity as well as the induction of apoptosis, inhibition of proliferation, and induction of differentiation. Because Michael acceptor groups had previously been identified as critical for the induction of cytoprotective enzymes[80,81], triterpenoid activity in suppressing inflammation was compared to their activity in inducing cytoprotective enzymes[82]. These studies show that triterpenoids are very potent inducers of cytoprotective NQO1 enzyme activity in vitro. However, triterpenoid analogs do not induce NQO1 or suppress nitric oxide production in Nrf2-deficient or Keap1-deficient mouse embryonic fibroblasts, indicating that this activity occurs through Keap1–Nrf2 signaling[82]. CDDO-Im and the parent molecule CDDO also induce cytoprotective heme oxygenase-1 in vitro and in vivo[83]. Low concentrations of CDDO-Im also reduce reactive oxygen species formation in U937 cells challenged with tert-butyl hydroperoxide. However, this cytoprotective activity is lost in Nrf2-deficient cells[83].
Due to the impressive potency of CDDO-Im noted in the studies described earlier, further studies were conducted to assess its chemopreventive potential in vivo. Microarray studies in wild-type and Nrf2-deficient mice showed that CDDO-Im induces many hepatic cytoprotective genes in a Nrf2-dependent manner. Furthermore, CDDO-Im is an extremely potent chemopreventive agent against aflatoxin-induced hepatic tumorigenesis in rats[14]. CDDO-Im reduces the hepatic burden of preneoplastic foci by 85% at the lowest dose of 1 µmol/kg body weight and more than 99% at the highest dose of 100 µmol/kg body weight. CDDO-Im inhibits aflatoxin-DNA adduct formation by 40%–90% over the range of 1–100 µmol/kg body weight. In addition, CDDO-Im increases RNA transcripts of aflatoxin metabolism genes, including Gsta2 and Gsta5, following an oral dose of 1 µmol/kg body weight[14]. CDDO-Im is approximately 100-fold more potent than oltipraz in this rat antitumorigenesis model.
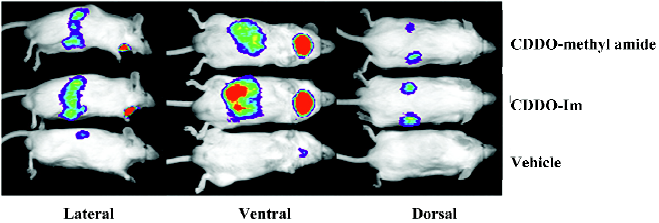
The pharmacodynamic action of synthetic triterpenoids has been further evaluated in vivo. The activation of Keap1–Nrf2–ARE signaling has been shown using 2 lines of transgenic reporter mice. Transgenic mice with the ARE of Nqo1 linked to a luciferase reporter can be used to identify both the localization and extent of ARE activation using in vivo bioluminescence. When Nqo1–ARE–luciferase mice are treated with triterpenoids, luciferase reporter gene expression is induced in the kidneys, salivary gland, liver, and intestines (Figure 4)[84]. A more precise localization of pharmacodynamic action can be obtained using Nqo1–ARE–human placental alkaline phosphatase (hPlap) reporter mice. These mice carry the ARE of Nqo1 linked to an hPlap reporter gene which can be detected by immunohistochemistry[85]. Nqo1–ARE–hPlap reporter gene expression is increased in hepatocytes following treatment with CDDO-Im[84]. Furthermore, several triterpenoid analogs may be effective chemopreventive agents in multiple target tissues as indicated by potent induction of Nrf2-regulated cytopro-tective genes in the liver, lung, small intestine mucosa, and brain[84].
While other analogs have not been studied against aflatoxin-induced hepatic tumorigenesis, the methyl ester and ethyl amide derivatives (called CDDO-ME and CDDO-EA, respectively) have been evaluated in a post-initiation lung cancer mouse model. As suggested by their ability to induce Nrf2-regulated cytoprotective genes in the lung[84], CDDO-ME and CDDO-EA are effective chemopreventive agents in the lung. CDDO-ME and CDDO-EA reduce the number, size, and severity of tumors on the surface of the lungs[86]. This study also has important implications for cancer chemoprevention in other organ sites. Post-initiation chemoprevention had not previously been evaluated with these compounds and is of critical importance because most interventions for chemoprevention would occur over a long period of time.
Human trials
The evaluation of potential chemopreventive agents in rodents has provided a wealth of information on which human clinical trials have been developed. The animal studies described earlier have identified agents with the greatest chemopreventive potential. Furthermore, rodent studies have elucidated the mechanisms of aflatoxin carcinogenesis and validated critical biomarkers for use in human studies.
Oltipraz Clinical trials have shown that oltipraz modulates the activities of both conjugating/detoxication enzymes as well as cytochrome P450s. A single 125 mg oral dose of oltipraz reduced CYP1a2 activity by 75% in healthy individuals[87]. Similar doses also increased GST activity in peripheral lymphocytes[88]. A dose-finding study using 125, 250, 500, or 1000 mg/m2 oltipraz showed increased GST activity in peripheral mononuclear cells and colon mucosa biopsies only at the lower doses[89]. Nqo1 RNA transcripts were also increased at 250 mg/m2[89]. Together, these studies confirm that oltipraz increases cytoprotective enzymes in humans.
Phase IIa intervention trials evaluated modulation of carcinogen metabolism following treatment with oltipraz. Participants for this randomized, placebo-controlled, double-blind study were recruited from Daxin township, Qidong, China. These residents have high dietary exposure to aflatoxins as well as a high risk of HCC. Two hundred forty adults with good general health and detectable serum aflatoxin–albumin adduct levels were randomized to receive placebo, 125 mg oltipraz administered daily or 500 mg oltipraz administered weekly. Urine samples were collected at 2 week intervals during the 8 week intervention period and during an 8 week follow-up period. Urine samples collected after the first month of intervention were assayed for aflatoxin metabolites[17]. These samples were evaluated for alterations in the activation product, aflatoxin M1, and the detoxication product, aflatoxin–mercapturic acid. After 1 month of weekly doses of 500 mg oltipraz, the level of aflatoxin M1 excreted in the urine was decreased by 51%. However, the aflatoxin-mercapturic acid levels were not significantly altered. Poten-tial modulation of detoxication enzymes may be masked by inhibition of the activation of aflatoxin B1. The daily administration of 125 mg oltipraz increased aflatoxin–mercapturic acid excretion 2.6-fold, but with only a modest effect on aflatoxin M1 excretion. This trial shows that induction of cytoprotective genes can be translated into the modulation of aflatoxin disposition in humans.
Broccoli sprouts Broccoli sprouts contain an abundance of glucosinolates and isothiocyanates, making them an attractive food-based candidate for chemoprevention. Clinical studies have evaluated metabolism, safety, tolerance, and biomarkers of carcinogenesis using broccoli sprouts. Evaluation of broccoli sprout preparations has shown that isothiocyanates are approximately 6 times more bioavailable than the precursor glucosinolates[90]. A placebo-controlled, double-blind, randomized phase I clinical study evaluated broccoli sprout preparations containing either glucosinolates or isothiocyanates (principally sulforaphane)[91]. The treatment groups received doses of 25 µmol glucosinolates, 100 µmol glucosinolates, or 25 µmol isothiocyanate. No significant or consistent toxicities were observed with any of the broccoli sprout preparations[91]. Interventions using hot water infusions of broccoli sprouts were evaluated in residents of Qidong, China[92]. Modulation of the disposition of aflatoxin was evaluated. Two hundred healthy adults drank infusions of either 400 µmol glucoraphanin or a placebo beverage nightly for 2 weeks. Again, no problems with safety or tolerance were observed. Urinary aflatoxin-DNA adducts were not different between the 2 interventions. However, measurement of urinary dithiocarbamate levels (sulforaphane metabolites) showed interindividual differences in bioavail-ability. Further analysis to control for the bioavailability of sulforaphane showed a highly significant inverse association between the levels of dithiocarbamates excreted and aflatoxin-DNA adducts[92]. The reduction of aflatoxin-DNA adducts is most likely due to induction of GST activity by sulforaphane. This study shows that aflatoxin disposition can be altered by the administration of glucosinolate-rich broccoli sprout preparations. A parallel inverse association was observed with the elimination of phenanthrene tetraols, demonstrating that the metabolism of polycyclic aromatic hydrocarbons can also be modulated[92]. Future studies with broccoli sprouts will require preparations which produce a higher yield and consistent level of sulforaphane bioavailability.
Triterpenoids CDDO and CDDO-ME are currently in clinical development by Reata Pharmaceuticals (Dallas, TX,USA). CDDO is in phase 1 studies in patients with relapsed and refractory leukemia. It is also being evaluated in patients with solid tumors. CDDO-ME is in phase 1 clinical development in patients with solid tumors and lymphoid malignancies[93]. While this class of synthetic triterpenoids is not yet in clinical trials for chemoprevention, the clinical trials for chemotherapy could provide critical data in humans to facilitate chemoprevention studies in the future.
Future directions
The clinical development of cancer prevention has been hampered by concerns about safety and liability, as well as few resources directed towards cancer prevention. The design of clinical cancer prevention trials must address these challenges efficiently. Clinical trials which evaluate disease as the end-point may not always be the most appropriate means to translate promising preclinical chemoprevention strategies into the clinic. These very costly trials require large numbers of participants and long-term follow up. “Phase 0” trials may be a more efficient strategy to initiate the translation of chemopreventive agents into the clinic. Phase 0 trials have been suggested as a way to improve the speed and success of chemotherapeutic drug development[94], but this approach is also relevant to chemoprevention. Exploratory phase 0 trials can provide proof of concept, determine the feasibility of assays for target modulation, evaluate biomarker modulation, and provide pharmacokinetic data to provide information for the subsequent development of investigational chemopreventive agents. As these end-points require fewer participants, short-term follow up, and require participants to receive limited doses of chemopreventive agents, this minimizes the risk of toxicity, reduces cost, and can provide earlier feedback on the pharmacodynamic activity of test agents in humans. However, phase 0 trials are only practicable for agents where the mechanism of action is well understood. Furthermore, phase 0 trials for cancer chemoprevention require that biomarkers for the targeted carcinogenic pathway are well validated. The utility of this approach has already been shown in trials using broccoli sprouts[90–92]. These studies provided proof of concept showing that aflatoxin disposition can be altered by the administration of glucosinolate-rich broccoli sprout preparations. In addition, the pharmacokinetic data collected during these studies identified the need for methods to achieve a consistent level of sulforaphane bioavailability in the broccoli sprout preparations. This information will be critical in designing further broccoli sprout interventions.
While research and resources are currently focused on cancer chemotherapy, chemoprevention is a practical strategy to reduce the incidence of HCC as well as many other types of cancer. Induction of Nrf2-regulated cytoprotective response has been proven to be a successful chemopreventive strategy in many animal models and has shown promise in clinical trials. There are many challenges for the clinical development of cancer chemopreventive agents, but success will have tremendous impact on the incidence of cancer development.
Acknowledgements
We would like to thank our colleagues both past and present for their contributions to our studies on chemo-prevention of liver cancer.
References
- Kensler TW, Egner PA, Wang JB, Zhu YR, Zhang BC, Lu PX, et al. Chemoprevention of hepatocellular carcinoma in aflatoxin endemic areas. Gastroenterology 2004;127:S310-8.
- Chen JG, Parkin DM, Chen QG, Lu JH, Shen QJ, Zhang BC, et al. Screening for liver cancer: results of a randomised controlled trial in Qidong, China. J Med Screen 2003;10:204-9.
- Chang MH, Chen CJ, Lai MS, Hsu HM, Wu TC, Kong MS, et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med 1997;336:1855-9.
- Kensler TW, Qian GS, Chen JG, Groopman JD. Translational strategies for cancer prevention in liver. Nat Rev Cancer 2003;3:321-9.
- Eaton DL, Gallagher EP. Mechanisms of aflatoxin carcinogenesis. Annu Rev Pharmacol Toxicol 1994;34:135-72.
- Ishii K, Maeda K, Kamataki T, Kato R. Mutagenic activation of aflatoxin B1 by several forms of purified cytochrome P-450. Mutat Res 1986;174:85-8.
- Bennett RA, Essigmann JM, Wogan GN. Excretion of an aflatoxin-guanine adduct in the urine of aflatoxin B1-treated rats. Cancer Res 1981;41:650-4.
- Sabbioni G, Skipper PL, Buchi G, Tannenbaum SR. Isolation and characterization of the major serum albumin adduct formed by aflatoxin B1 in vivo in rats. Carcinogenesis 1987;8:819-24.
- Scholl PF, Musser SM, Groopman JD. Synthesis and characterization of aflatoxin B1 mercapturic acids and their identification in rat urine. Chem Res Toxicol 1997;10:1144-51.
- Wild CP, Hudson GJ, Sabbioni G, Chapot B, Hall AJ, Wogan GN, et al. Dietary intake of aflatoxins and the level of albumin-bound aflatoxin in peripheral blood in the Gambia, West Africa. Cancer Epidemiol Biomarkers Prev 1992;1:229-34.
- Groopman JD, Zhu JQ, Donahue PR, Pikul A, Zhang LS, Chen JS, et al. Molecular dosimetry of urinary aflatoxins-DNA adducts in people living in Guangxi autonomous region, People’s Republic of China. Cancer Res 1992;52:45-52.
- Groopman JD, Wild CP, Hasler J, Junshi C, Wogan GN, Kensler TW. Molecular epidemiology of aflatoxin exposures: validation of aflatoxin-N7-guanine levels in urine as a biomarker in experimental rat models and humans. Environ Health Perspect 1993;99:107-13.
- Groopman JD, Hall AJ, Whittle H, Hudson GJ, Wogan GN, Montesano R, et al. Molecular dosimetry of aflatoxin-N7-guanine in human urine obtained in the Gambia, West Africa. Cancer Epidemiol Biomarkers Prev 1992;1:221-7.
- Yates MS, Kwak MK, Egner PA, Groopman JD, Bodreddigari S, Sutter TR, et al. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res 2006;66:2488-94.
- Kensler TW, Egner PA, Davidson NE, Roebuck BD, Pikul A, Groopman JD. Modulation of aflatoxin metabolism, aflatoxin-N7-guanine formation, and hepatic tumorigenesis in rats fed ethoxyquin: role of induction of glutathione S-transferases. Cancer Res 1986;46:3924-31.
- Groopman JD, Donahue PR, Zhu JQ, Chen JS, Wogan GN. Aflatoxin metabolism in humans: detection of metabolites and nucleic acid adducts in urine by affinity chromatography. Proc Natl Acad Sci USA 1985;82:6492-6.
- Wang JS, Shen X, He X, Zhu YR, Zhang BC, Wang JB, et al. Protective alterations in phase 1 and 2 metabolism of aflatoxin B1 by oltipraz in residents of Qidong, People’s Republic of China. J Natl Cancer Inst 1999;91:347-54.
- Scholl P, Musser SM, Kensler TW, Groopman JD. Inhibition of aflatoxin Ml excretion in rat urine during dietary intervention with oltipraz. Carcinogenesis 1996;17:1385-8.
- Qian GS, Ross RK, Yu MC, Yuan JM, Gao YT, Henderson BE, et al. A follow-up study of urinary markers of aflatoxin exposure and liver cancer risk in Shanghai, People’s Republic of China. Cancer Epidemiol Biomarkers Prev 1994;3:3-10.
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci USA 2001;98:3410-5.
- McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, et al. The Cap’n’collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res 2001;61:3299-307.
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997;236:313-22.
- Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J 2002;365:405-16.
- Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci USA 1999; 96: 12 731–6.
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 2002;62:5196-203.
- Osburn WO, Wakabayashi N, Misra V, Nilles T, Biswal S, Trush MA, et al. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch Biochem Biophys 2006;454:7-15.
- Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemo-preventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem 2003;278:8135-45.
- Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dithiole-3-thione. Mol Med 2001;7:135-45.
- Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol 2001;173:154-60.
- Kwak MK, Egner PA, Dolan PM, Ramos-Gomez M, Groopman JD, Itoh K, et al. Role of phase 2 enzyme induction in chemo-protection by dithiolethiones. Mutat Res 2001;480–481:305-15.
- Ramos-Gomez M, Dolan PM, Itoh K, Yamamoto M, Kensler TW. Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene–DNA adducts and tumor yield in mice. Carcinogenesis 2003;24:461-7.
- Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res 2006;66:8293-6.
- Telakowski-Hopkins CA, King RG, Pickett CB. Glutathione S-transferase Ya subunit gene: identification of regulatory elements required for basal level and inducible expression. Proc Natl Acad Sci USA 1988;85:1000-4.
- Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem 1990;265:14648-53.
- Moinova HR, Mulcahy RT. An electrophile responsive element (EpRE) regulates beta-naphthoflavone induction of the human gamma-glutamylcysteine synthetase regulatory subunit gene. Constitutive expression is mediated by an adjacent AP-1 site. J Biol Chem 1998;273:14683-9.
- Li Y, Jaiswal AK. Regulation of human NAD(P)H:quinone oxidoreductase gene. Role of AP1 binding site contained within human antioxidant response element. J Biol Chem 1992;267:15097-104.
- Favreau LV, Pickett CB. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J Biol Chem 1991;266:4556-61.
- Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci USA 1993;90:2965-9.
- Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of Keap1 in cellular protective responses. Chem Res Toxicol 2005;18:1779-91.
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999;13:76-86.
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 2004;24:7130-9.
- Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol 2005;25:162-71.
- Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol 2004;24:8477-86.
- McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem 2003;278:21592-600.
- Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA 2002;99:11908-13.
- Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, et al. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol 2006;26:221-9.
- Yu R, Tan TH, Kong AT. Butylated hydroxyanisole and its metabolite tert-butylhydroquinone differentially regulate mitogen-activated protein kinases. The role of oxidative stress in the activation of mitogen-activated protein kinases by phenolic antioxidants. J Biol Chem 1997;272:28962-70.
- Yu R, Lei W, Mandlekar S, Weber MJ, Der CJ, Wu J, et al. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J Biol Chem 1999;274:27545-52.
- Yu R, Chen C, Mo YY, Hebbar V, Owuor ED, Tan TH, et al. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism. J Biol Chem 2000;275:39907-13.
- Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem 2002;277:42769-74.
- Lee JM, Hanson JM, Chu WA, Johnson JA. Phosphatidylinositol 3-kinase, not extracellular signal-regulated kinase, regulates activation of the antioxidant-responsive element in IMR-32 human neuroblastoma cells. J Biol Chem 2001; 276: 20 011–6.
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 2003;23:7198-209.
- Kensler TW, Egner PA, Trush MA, Bueding E, Groopman JD. Modification of aflatoxin B1 binding to DNA in vivo in rats fed phenolic antioxidants, ethoxyquin and a dithiothione. Carcinogenesis 1985;6:759-63.
- Wattenberg LW, Lam LT. Inhibition of chemical carcinogenesis by phenols, coumarins, aromatic isothiocyanates, flavones, and indoles. In: Zedeck MS, Lipkin M, editors. Inhibition of tumor induction and development. New York: Plenum Press; 1981. p1–22.
- Cabral JR, Neal GE. The inhibitory effects of ethoxyquin on the carcinogenic action of aflatoxin B1 in rats. Cancer Lett 1983;19:125-32.
- Nair S, Xu C, Shen G, Hebbar V, Gopalakrishnan A, Hu R, et al. Pharmacogenomics of phenolic antioxidant butylated hydroxyanisole (BHA) in the small intestine and liver of Nrf2 knockout and C57BL/6J mice. Pharm Res 2006;23:2621-37.
- Hayes JD, Chanas SA, Henderson CJ, McMahon M, Sun C, Moffat GJ, et al. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem Soc Trans 2000;28:33-41.
- Ansher SS, Dolan P, Bueding E. Biochemical effects of dithiolthiones. Food Chem Toxicol 1986;24:405-15.
- Kensler TW, Egner PA, Dolan PM, Groopman JD, Roebuck BD. Mechanism of protection against aflatoxin tumorigenicity in rats fed 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione (oltipraz) and related 1,2-dithiol-3-thiones and 1,2-dithiol-3-ones. Cancer Res 1987;47:4271-7.
- Roebuck BD, Liu YL, Rogers AE, Groopman JD, Kensler TW. Protection against aflatoxin B1-induced hepatocarcinogenesis in F344 rats by 5-(2-pyrazinyl)-4-methyl-1,2-dithiole-3-thione (oltipraz): predictive role for short-term molecular dosimetry. Cancer Res 1991;51:5501-6.
- Dimitrov NV, Bennett JL, McMillan J, Perloff M, Leece CM, Malone W. Clinical pharmacology studies of oltipraz—a potential chemopreventive agent. Invest New Drugs 1992;10:289-98.
- Roebuck BD, Curphey TJ, Li Y, Baumgartner KJ, Bodreddigari S, Yan J, et al. Evaluation of the cancer chemopreventive potency of dithiolethione analogs of oltipraz. Carcinogenesis 2003;24:1919-28.
- Pietsch EC, Chan JY, Torti FM, Torti SV. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J Biol Chem 2003;278:2361-9.
- Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res 2004;64:6424-31.
- Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, et al. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (–/–) mice. Cancer Lett 2006;243:170-92.
- Gerhauser C, You M, Liu J, Moriarty RM, Hawthorne M, Mehta RG, et al. Cancer chemopreventive potential of sulforamate, a novel analogue of sulforaphane that induces phase 2 drug-metabolizing enzymes. Cancer Res 1997;57:272-8.
- Dinkova-Kostova AT, Fahey JW, Talalay P. Chemical structures of inducers of nicotinamide quinone oxidoreductase 1 (NQO1). Methods Enzymol 2004;382:423-48.
- Hong F, Freeman ML, Liebler DC. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol 2005;18:1917-26.
- Zhang Y, Kensler TW, Cho CG, Posner GH, Talalay P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc Natl Acad Sci USA 1994;91:3147-50.
- Singh GB, Singh S, Bani S, Gupta BD, Banerjee SK. Anti-inflammatory activity of oleanolic acid in rats and mice. J Pharm Pharmacol 1992;44:456-8.
- Nishino H, Nishino A, Takayasu J, Hasegawa T, Iwashima A, Hirabayashi K, et al. Inhibition of the tumor-promoting action of 12-O-tetradecanoylphorbol-13-acetate by some oleanane-type triterpenoid compounds. Cancer Res 1988;48:5210-5.
- Lapillonne H, Konopleva M, Tsao T, Gold D, McQueen T, Sutherland RL, et al. Activation of peroxisome proliferator-activated receptor gamma by a novel synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces growth arrest and apoptosis in breast cancer cells. Cancer Res 2003;63:5926-39.
- Konopleva M, Tsao T, Estrov Z, Lee RM, Wang RY, Jackson CE, et al. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Res 2004;64:7927-35.
- Ito Y, Pandey P, Place A, Sporn MB, Gribble GW, Honda T, et al. The novel triterpenoid 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid induces apoptosis of human myeloid leukemia cells by a caspase-8-dependent mechanism. Cell Growth Differ 2000;11:261-7.
- Samudio I, Konopleva M, Hail N Jr, Shi YX, McQueen T, Hsu T, et al. 2-Cyano-3,12-dioxooleana-1,9-dien-28-imidazolide (CDDO-Im) directly targets mitochondrial glutathione to induce apoptosis in pancreatic cancer. J Biol Chem 2005;280:36273-82.
- Place AE, Suh N, Williams CR, Risingsong R, Honda T, Honda Y, et al. The novel synthetic triterpenoid, CDDO-imidazolide, inhibits inflammatory response and tumor growth in vivo. Clin Cancer Res 2003;9:2798-806.
- Honda T, Rounds BV, Bore L, Finlay HJ, Favaloro FG Jr, Suh N, et al. Synthetic oleanane and ursane triterpenoids with modified rings A and C: a series of highly active inhibitors of nitric oxide production in mouse macrophages. J Med Chem 2000;43:4233-46.
- Honda T, Honda Y, Favaloro FG Jr, Gribble GW, Suh N, Place AE, et al. A novel dicyanotriterpenoid, 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-onitrile, active at picomolar concentrations for inhibition of nitric oxide production. Bioorg Med Chem Lett 2002;12:1027-30.
- Honda T, Gribble GW, Suh N, Finlay HJ, Rounds BV, Bore L, et al. Novel synthetic oleanane and ursane triterpenoids with various enone functionalities in ring A as inhibitors of nitric oxide production in mouse macrophages. J Med Chem 2000;43:1866-77.
- Talalay P, De Long MJ, Prochaska HJ. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc Natl Acad Sci USA 1988;85:8261-5.
- Dinkova-Kostova AT, Massiah MA, Bozak RE, Hicks RJ, Talalay P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc Natl Acad Sci USA 2001;98:3404-9.
- Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, Suh N, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci USA 2005;102:4584-9.
- Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, et al. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res 2005;65:4789-98.
- Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther 2007;6:154-62.
- Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem 2002;81:1233-41.
- Liby K, Royce DB, Williams CR, Risingsong R, Yore MM, Honda T, et al. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res 2007;67:2414-9.
- Sofowora GG, Choo EF, Mayo G, Shyr Y, Wilkinson GR. In vivo inhibition of human CYP1A2 activity by oltipraz. Cancer Chemother Pharmacol 2001;47:505-10.
- Gupta E, Olopade OI, Ratain MJ, Mick R, Baker TM, Berezin FK, et al. Pharmacokinetics and pharmacodynamics of oltipraz as a chemopreventive agent. Clin Cancer Res 1995;1:1133-8.
- O’Dwyer PJ, Szarka CE, Yao KS, Halbherr TC, Pfeiffer GR, Green F, et al. Modulation of gene expression in subjects at risk for colorectal cancer by the chemopreventive dithiolethione oltipraz. J Clin Invest 1996;98:1210-7.
- Shapiro TA, Fahey JW, Wade KL, Stephenson KK, Talalay P. Chemoprotective glucosinolates and isothiocyanates of broccoli sprouts: metabolism and excretion in humans. Cancer Epidemiol Biomarkers Prev 2001;10:501-8.
- Shapiro TA, Fahey JW, Dinkova-Kostova AT, Holtzclaw WD, Stephenson KK, Wade KL, et al. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: a clinical phase I study. Nutr Cancer 2006;55:53-62.
- Kensler TW, Chen JG, Egner PA, Fahey JW, Jacobson LP, Stephenson KK, et al. Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxins–DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People’s Republic of China. Cancer Epidemiol Bio-markers Prev 2005;14:2605-13.
- Synthetic Triterpenoids for Treating Hematological Cancers, Solid Tumors, and Treatment-Related Toxicities [homepage on the Internet]. Dallas: Reata Pharmaceuticals, Inc; c2006. Available from: http://www.reatapharma.com/clinicalprograms.asp
- Kummar S, Kinders R, Rubinstein L, Parchment RE, Murgo AJ, Collins J, et al. Compressing drug development timelines in oncology using phase ‘0’ trials. Nat Rev Cancer 2007;7:131-9.
- Williams GM, Iatropoulos MJ. Inhibition of the hepatocarcino-genicity of aflatoxin B1 in rats by low levels of the phenolic antioxidants butylated hydroxyanisole and butylated hydroxyto-luene. Cancer Lett 1996;104:49-53.
- De Long MJ, Prochaska HJ, Talalay P. Induction of NAD(P)H:quinone reductase in murine hepatoma cells by phenolic anti-oxidants, azo dyes, and other chemoprotectors: a model system for the study of anticarcinogens. Proc Natl Acad Sci USA 1986;83:787-91.
- Egner PA, Kensler TW, Prestera T, Talalay P, Libby AH, Joyner HH, et al. Regulation of phase 2 enzyme induction by oltipraz and other dithiolethiones. Carcinogenesis 1994;15:177-81.
- Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci USA 1992;89:2399-403.
- Fahey JW, Dinkova-Kostova AT, Stephenson KK, Talalay P. The “Prochaska” microtiter plate bioassay for inducers of NQO1. Methods Enzymol 2004;382:243-58.