
Synergy of gemcitabine and lidamycin associated with NF-κB downregulation in pancreatic carcinoma cells1
Introduction
Pancreatic cancer is a highly-malignant neoplasm characterized by locally-advanced, unresectable disease or metastasis at the time of diagnosis. Over the last three decades, the 5-year survival of patients with pancreatic cancer has been <5% without significant improvement[1]. This poor prognosis is attributed to the high incidence of metastatic disease at diagnosis. Consequently, major improvement in the outlook of this disease will depend on the development of more effective drug therapies. Systemic therapy with gemcitabine (2´,2´-difluorodeoxycytidine), a deoxycytidine analog used for pancreatic cancer, has not increased the median survival of patients beyond 6 months, and often leads to resistance[2]. Therefore, an effective treatment modality for this devastating disease is urgently needed.
The potent antitumor activity of enediyne antibiotics has been the focus of attention because of their unique ability to damage the DNA of tumor cells by inducing single-strand and/or double-strand breaks through free radical attacks on the deoxyribose moieties in DNA[3]. Lidamycin (LDM; also named C-1027) is a member of the enediyne antibiotic family, which was produced by a Streptomyces globisporus C-1027 strain isolated in China[4,5]. The LDM molecule contains an enediyne chromophore responsible for the extremely potent bioactivity and a non-covalently-bound apoprotein, which forms a hydrophobic pocket for protecting the chromophore[6,7]. LDM shows extremely potent cytotoxicity, anti-angiogenic activity, and marked growth inhibition of transplantable tumors in mice[8–11]. It is currently being evaluated in phase II clinical trials as a potential chemotherapeutic agent in China.
Our previous study showed that LDM was highly active in targeting the Akt/NF-κB signal pathway and induced apoptosis and cell cycle arrest in human pancreatic cancer cells. Moreover, LDM could suppress the growth of xenografts in athymic nude mice[12]. In the present study, we found that LDM could potentiate the growth inhibition induced by gemcitabine in human pancreatic cancer cells, and that the synergy might be associated with NF-κB downregulation.
Materials and methods
Cells, reagents, and drugs Human pancreatic cancer cell lines PANC-1 and SW1990 were maintained in culture with Dulbecco’s modified Eagle’s medium (DMEM; Gibco BRL, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (Sigma, St Louis, MO, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. LDM was provided by Professor Lian-fang JIN from the Institute of Medicinal Biotechnology (Chinese Academy of Medical Sciences, Beijing, China). Gemcitabine was the product of Lilly France SA (Lille, France).
Cell proliferation assay The PANC-1 and SW1990 cells were plated in triplicate in a 96-well plate with 3000 cells/well and 4000 cells/well, respectively. After overnight incubation, the triplicate wells were treated with gemcitabine, LDM, and a combination of both for 48 h. The effects on cell growth were examined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In brief, 20 μL MTT solution (5 mg/mL in phosphate-buffered saline [PBS]; Sigma, USA) were added to each well and incubated for 4 h at 37 °C. The MTT formazan was dissolved in 150 μL DMSO and absorbance was measured by a microplate reader (Multiskan MK3, Thermo Labsystem, USA) at a wavelength of 570 nm.
In order to calculate the coefficient of drug interaction (CDI), the following equation was used:
CDI=AB/A×B,
where AB is the cell survival ratio of the combination group, A is the cell survival ratio of the LDM group, and B is the cell survival ratio of the gemcitabine group. CDI <1 indicates the synergistic effect of the drugs.
Analysis of apoptosis by terminal uridine deoxynucleotidyl transferase dUTP nick end labeling The nuclear DNA fragmentation of apoptotic cells was measured by TUNEL assay (DeadEnd colorimetric TUNEL system, Promega Madison, USA). Briefly, the cells were harvested, washed in PBS, resuspended in PBS, and added to the poly-lysine-coated slides. The cells were then fixed in 4% paraformaldehyde, permeabilized in 0.2% Triton X-100, and incubated with terminal deoxynucleotidyl transferase incubation buffer for 60 min in a 37 °C humidified chamber for 3´-OH labeling. The cells that bound with streptavidin horseradish peroxidase (HRP) and were stained with 3,3´-diaminobenzidine-tetrachloride were analyzed using a fluorescence microscope.
Fluorescein-isothiocyanate-Annexin V/propidium iodide apoptosis assay The cells were harvested and resuspended in 200 μL binding buffer. Then 10 μL fluorescein-isothiocyanate (FITC)-labeled, enhanced Annexin V (Baosai Biotechnology, Beijing, China) and 100 ng of propidium iodide (PI) were added. After incubation in the dark (15 min at room temperature or 30 min at 4 °C), the samples were diluted with 300 μL binding buffer. Flow cytometry was carried out on a FACScan instrument (Becton Dickinson, NY, USA), and the data were processed by WinMDI/PC software.
5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimida-zolylcarbocyanine iodide staining and confocal microscopy Following the experimental treatments, the cells seeded on 35 mm dishes were stained with JC-1 (Molecular Probes, Eugene, Oregon, USA) to determine the state of mitochondrial membrane potential. Briefly, the culture medium was removed from the adherent cells, and the monolayers were rinsed once with DMEM. The cell monolayers were incubated with DMEM containing 10% serum and 5 μg/mL JC-1 at 37 °C for 30 min. Then the cells were rinsed twice with DMEM, and images were obtained using a 10× objective on a confocal microscope (Leica SP2, Leica Microsystems, Wetzlar, German) excited at 488 nm (for JC-1) set to simultaneously detect green emissions (510–525 nm) and red emission (590 nm) channels using a dual band-pass filter.
Caspase-3 activity assay Caspase-3 activity was detected by the Apo-ONER homogeneous caspase-3/7 assay kit (Promega, USA). Briefly, the cells were seeded into 96-well plates (1×104/well). Following treatment with the drugs for 48 h, the cells were washed with ice-cold PBS. A total of 100 μL homogeneous caspase-3 reagent was added to each well. The contents were gently mixed and incubated for 4 h at room temperature in the dark. The fluorescence intensity of the Z-DEVD-R110 substrate was measured at an excitation wavelength of 498 nm and an emission wavelength of 521 nm using a microplate spectrofluorometer (Polarstar, BMG, Offenburg, Germany).
Western blotting analysis The total protein extract of the cells was prepared by incubation for 15 min on ice with an ice-cold hypotonic buffer containing 50 mmol/L Tris-HCl (pH7.5), 150 mmol/L NaCl, 2 mmol/L EDTA, 2 mmol/L ethyleneglycol bis(2-aminoethyl ether)tetraacetic acid (EGTA), 1 mmol/L dithiothreitol, 1% Nonidet P-40, 0.1% SDS, protease inhibitors (1 mmol/L phenylmethylsulfonyl fluoride, 5 mg/mL aprotinin, 5 mg/mL leupeptin, and 5 mg/mL pepstatin) and phosphatase inhibitors (20 mmol/L β-glycerophosphate, 50 mmol/L NaF, and 1 mmol/L Na3VO4). The lysates were centrifuged at 12 000×g for 12 min. Protein samples of an equal amount were denatured with 1 volume of 6× SDS sample buffer and loaded on SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The blots were blocked for 60 min at room temperature with 5% non-fat milk powder and 0.1% Tween-20 in PBS, and exposed overnight at 4 °C to a primary antibody against K-ras, NF-κB, and Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), respectively. The blots were washed with TBST (20 mmol/L Tris-HCl [pH 7.4], 150 mmol/L NaCl, and 0.1% Tween-20) for 5 min (3 times) and exposed for 60 min at room temperature to an appropriate HRP-linked secondary antibody (Santa Cruz Biotechnology, USA). Complexes of the primary and secondary antibodies were visualized using enhanced chemiluminescence Western blotting detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
RT-PCR Total cellular RNA was extracted from the cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the protocol recommended by the manufacturer. RT-PCR amplification was performed on the extracted RNA using the Superscript one-step RT-PCR kit (Invitrogen, USA) according to manufacturer’s instructions. The primer pair for K-ras was as follows: 5´-ACTTGTGGTAGTTGGAGCTG-3´ (sense) and 5´-CTAACAGTCTGCATGGAGC-3´ (antisense). Reverse transcription was performed at 55 ℃ for 15 min. The denaturation and amplification conditions were 95 °C for 30 s followed by up to 25 cycles of PCR. Each cycle of PCR included denaturation at 95 °C for 15 s, annealing at 55 °C for 30 s, and extension at 72 °C for 40 s. After PCR amplification, the fragments were analyzed by 2% agarose gel electrophoresis. GAPDH was used as a positive control. The sizes were estimated by comparison with molecular weight markers.
Statistical analysis The results were expressed as mean±SD. Treatment effects were compared using Student’s t-test, and differences between means were considered to be significant when P<0.05.
Results
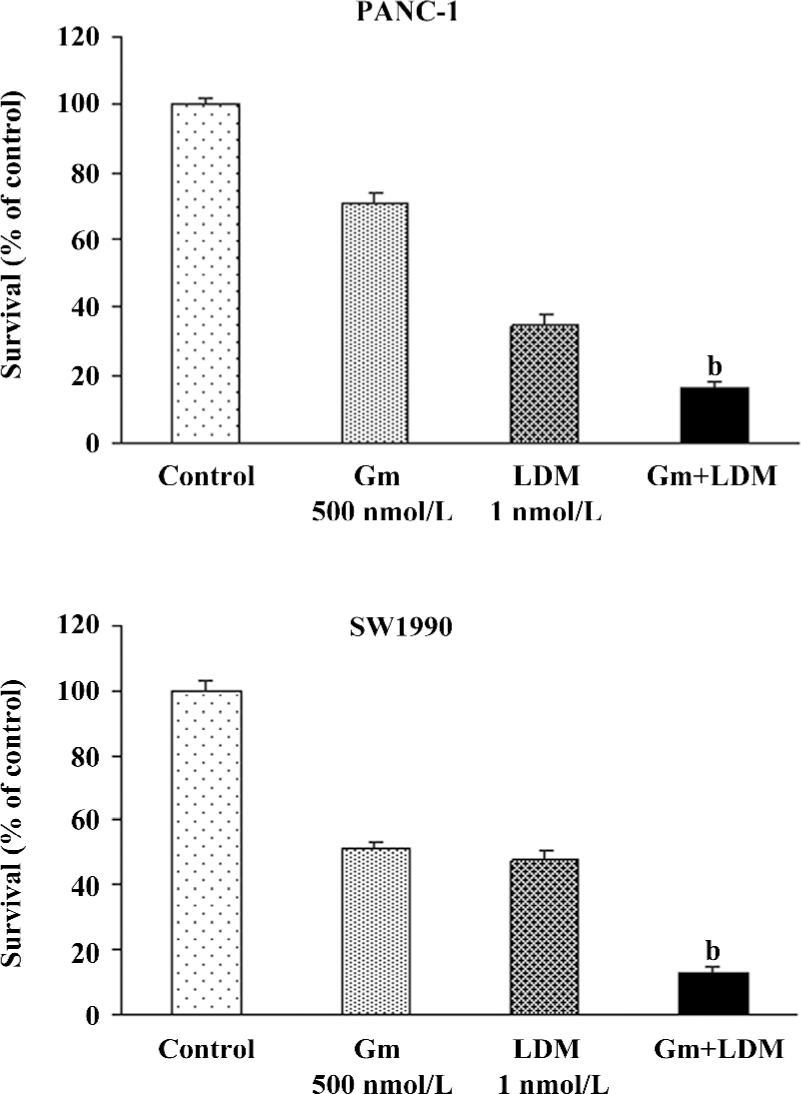
Gemcitabine-induced growth inhibition was potentiated by LDM in PANC-1 and SW1990 cells The growth of the PANC-1 and SW1990 cells treated with LDM (1 nmol/L), gemcitabine (500 nmol/L), or a combination of both was determined by MTT assay. The dose used in the present study was chosen based upon a preliminary dose escalation study. A significant reduction in growth was observed in both cells treated in combination compared with treatment with LDM or gemcitabine alone (Figure 1). The CDI was less than 0.7, which means that the 2 drugs have a synergistic effect.
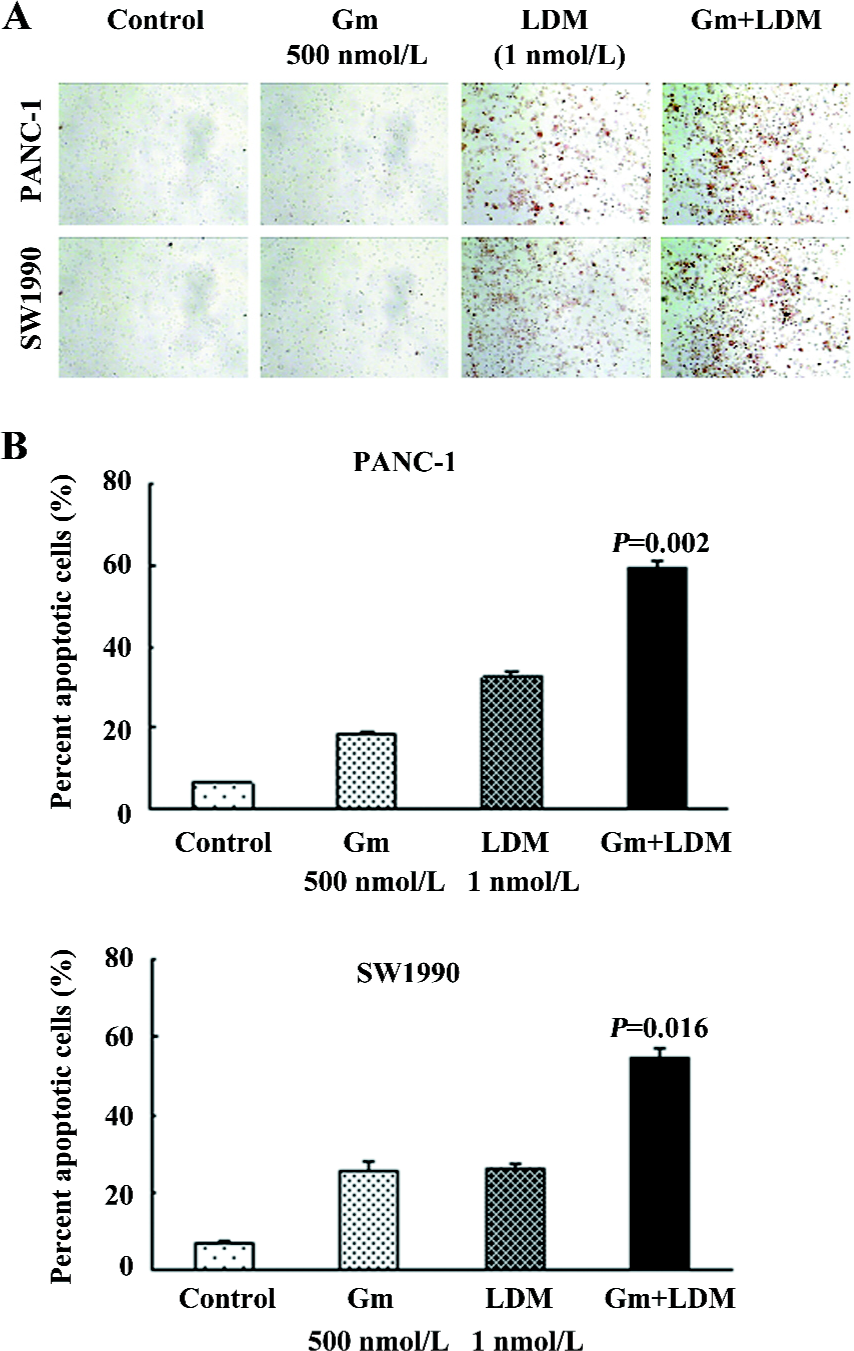
Gemcitabine-induced apoptosis was sensitized by LDM in PANC-1 and SW1990 cells We observed the induction of apoptosis in the pancreatic cancer cells treated with either gemcitabine, LDM, or a combination of both. Relative to single agents, the combination treatment induced greater apoptosis in both cell lines as shown by both the TUNEL analysis and flow cytometry combined with FITC-Annexin V/PI staining (Figure 2). The ratios of apoptosis were 59.44%±1.54% and 54.68%±2.62% in the combination groups, whereas those of the gemcitabine groups were 18.48%±0.94% and 25.79%±2.06% in the PANC-1 and SW1990 cells, respectively. These results are consistent with cell growth inhibition studies by MTT, suggesting that the loss of viable cells by LDM and gemcitabine is partly due to the induction of an apoptotic cell death mechanism.
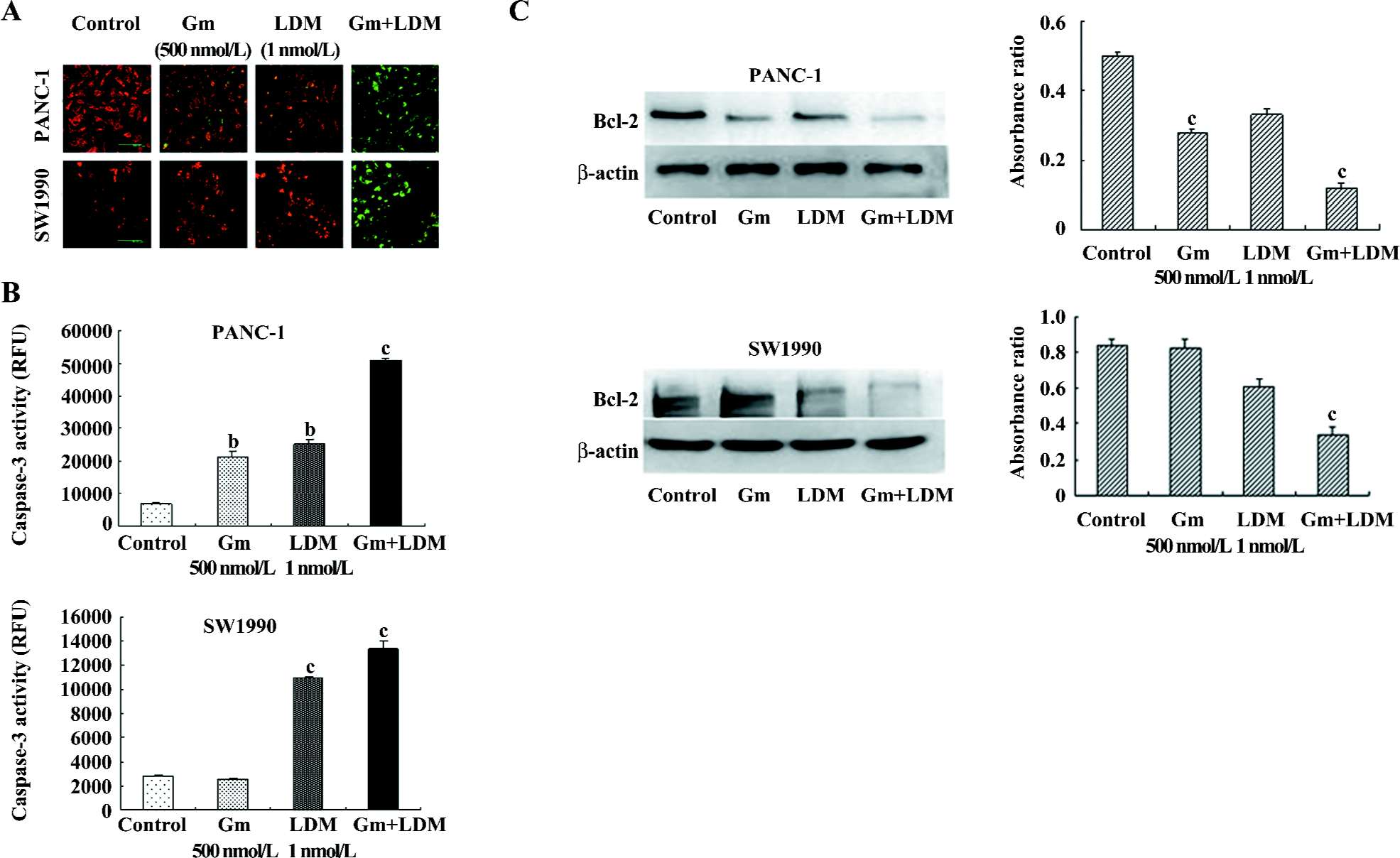
Gemcitabine-induced apoptosis signaling was augmented by LDM in PANC-1 and SW1990 cells In an attempt to explore the mechanism of enhanced apoptotic process induced by the treatment of cells with LDM and gemcitabine, we assessed the changes of mitochondrial membrane potential, caspase-3 activity, and the levels of Bcl-2 in the PANC-1 and SW1990 cells. Our results showed that the combination treatment could decrease mitochondrial membrane potential and enhance caspase-3 activity significantly. The results for the anti-apoptotic Bcl-2 proteins also showed downregulation in the combination group relative to the signal-agent treatment and untreated control (Figure 3).
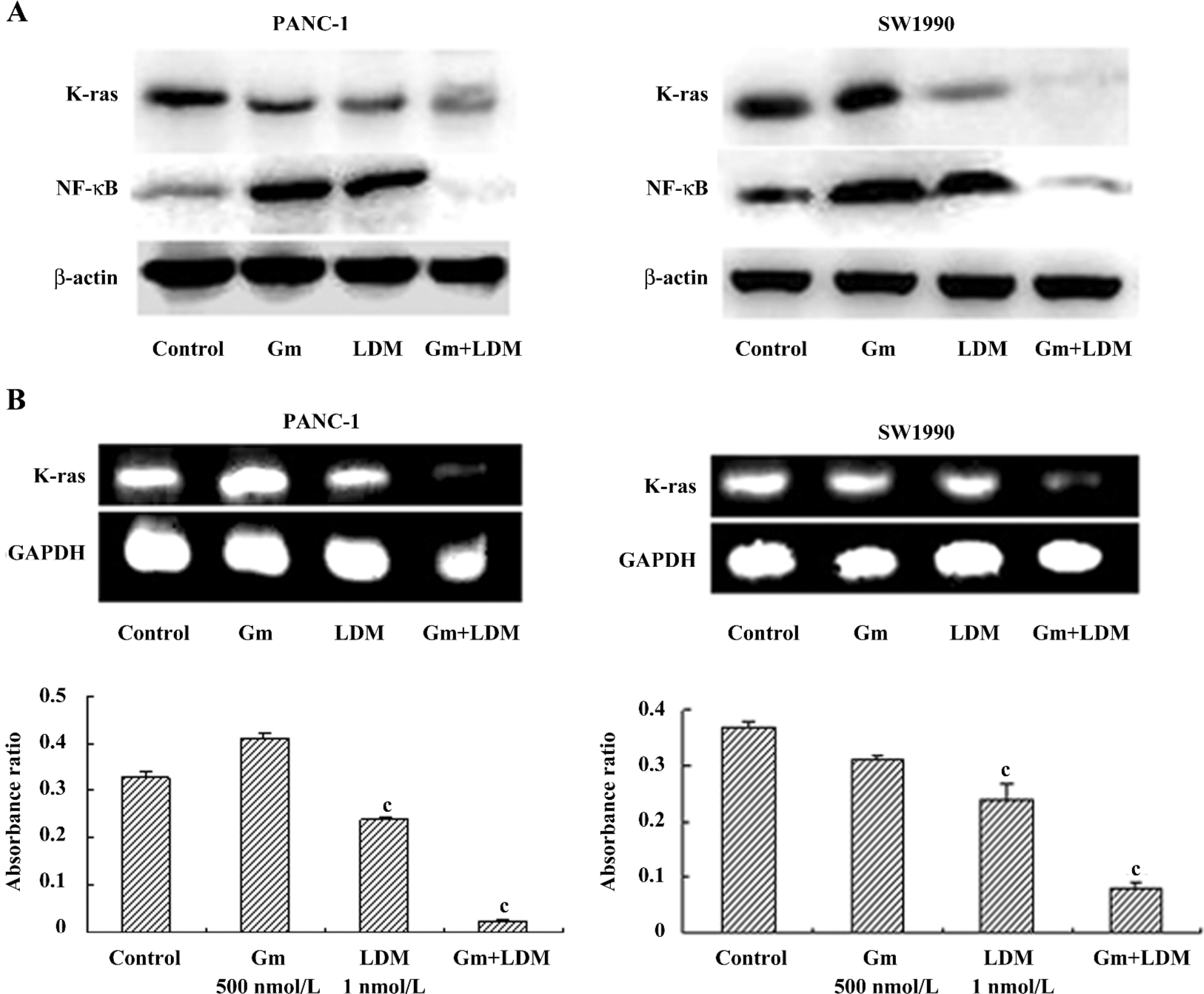
Effects of gemcitabine, LDM, and a combination on the K-ras/NF-κB signaling pathway. In the present study, we found that gemcitabine treatment enhanced NF-κB levels in PANC-1 and SW1990 cells. However, gemcitabine in combination with LDM prevented the gemcitabine-induced NF-κB enhancement through the inhibition of K-ras. Meanwhile, the levels of K-ras mRNA decreased in both cells after being treated with a combination of both (Figure 4).
Discussion
Pancreatic cancer is now one of the most common causes of cancer death worldwide. K-ras mutations are present in up to 90% of cases of pancreatic cancer[13]. The expression of mutant K-ras activates the protein kinase B pathway, resulting in the activation of the NF-κB transcriptional factor[14]. NF-κB has been shown to inhibit apoptosis in response to chemotherapeutic agents[15]. Compounds targeting the NF-κB pathway can sensitize pancreatic tumor cells by counteracting resistance mechanisms, and therefore, deserve further evaluation as in the chemotherapy and possible chemoprevention of pancreatic cancer[16,17]. In the present study, we found that gemcitabine in combination with LDM could prevent the gemcitabine-induced NF-κB enhancement through the inhibition of K-ras.
Highly-metabolically-active mitochondria were particularly sensitive and were vulnerable targets to cellular stress[18]. Membrane depolarization has been widely associated with the release of the apoptotic factor, cytochrome c, which amplifies pro-apoptotic caspase cascades, promoting cell death[18,19]. The anti-apoptotic properties of Bcl-2 and Bcl-xL have been attributed to their ability to prevent translocation of cytochrome c to the cytosol, and thus, interfere with the subsequent activation of cytosolic caspases and apoptosis[20,21]. In the present study, we found that LDM potentiated the gemcitabine-induced cell killing by reducing mitochondrial membrane potential and increasing the caspase-3 activity. The results for the anti-apoptotic Bcl-2 proteins also showed downregulation in the combination group relative to the signal-agent treatment and untreated control.
In conclusion, our current findings have shown a synergistic effect of gemcitabine and LDM in certain pancreatic cancer cell lines. The synergy was probably associated with NF-κB downregulation. Accordingly, further mechanistic studies would be useful in the treatment of patients with pancreatic carcinoma.
References
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al. Cancer statistics. Cancer J Clin 2006;56:106-30.
- Abbruzzese JL. New application of gemcitabine and future directions in the management of pancreatic cancer. Cancer 2002;95:941-5.
- Dziegielewski J, Beerman TA. Cellular responses to the DNA strand-scission enediyne C-1027 can be independent of ATM, ATR, and DNA-PK kinases. J Biol Chem 2002; 277: 20 549–54.
- Hu JL, Xue YC, Xie MY, Zhang R, Otani T, Minami Y, et al. A new macromolecular antitumor antibiotic, C-1027. I. Discovery, taxonomy of producing organism, fermentation and biological activity. J Antibiot 1988;41:1575-9.
- Otani T, Minami Y, Marunaka T, Zhang R, Xie MY. A new macromolecular antitumor antibiotic, C-1027. II. Isolation and physico-chemical properties. J Antibiot 1988;41:1580-5.
- Sakata N, Ikeno S, Hori M, Hamada M, Otani T. Cloning and nucleotide sequencing of the antitumor antibiotic C-1027 apoprotein gene. Biosci Biotechnol Biochem 1992;56:1592-5.
- Tanaka T, Hirama M, Otani T. Solution structures of C-1027 apoprotein and its complex with the aromatized chromophore. J Mol Biol 2001;309:267-83.
- Zhen YS, Ming XY, Yu B, Otani T, Saito H, Yamada Y. A new macromolecular antitumor antibiotic, C-1027. III. Antitumor activity. J Antibiot 1989;42:1294-8.
- Xu YJ, Zhen YS, Goldberg IH. C-1027 chromophore, a potent new enediyne antitumor antibiotic, induces sequence-specific double-strand DNA cleavage. Biochemistry 1994;33:5947-54.
- Zhen HY, Xue YC, Zhen YS. Inhibition of angiogenesis by antitumor antibiotic C-1027 and its effect on tumor metastasis. Natl Med J China 1997;77:657-60.
- Huang YH, Shang BY, Zhen YS. Antitumor efficacy of lidamycin on hepatoma and active moiety of its molecule. World J Gastroenterol 2005;11:3980-4.
- Chen J, Ou-Yang ZG, Zhang SH, Zhen YS. Down-regulation of NF-κB by lidamycin in association with inducing apoptosis in human pancreatic cancer cells and inhibiting xenograft growth. Oncol Rep 2007;17:1445-51.
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras gene. Cell 1988;53:549-54.
- Osada M, Tolkacheva T, Li W, Chan TO, Tsichlis PN, Saez R, et al. Differential roles of Akt, Rac, and Ral in R-Ras-mediated cellular transformation, adhesion, and survival. Mol Cell Biol 1999;19:6333-44.
- Arlt A, Gehrz A, Muerkoster S, Vorndamm J, Kruse ML, Folsch UR, et al. Role of NF-κB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 2003;22:3243-51.
- Mohammad RM, Banerjee S, Li Y, Aboukameel A, Kucuk O, Sarkar FH. Cisplatin-induced antitumor activity is potentiated by the soy isoflavone genistein in BxPC-3 pancreatic tumor xenografts. Cancer 2006;106:1260-8.
- Yip-Schneider MT, Nakshatri H, Sweeney CJ, Marshall MS, Wiebke EA, Schmidt CM. Parthenolide and sulindac cooperate to mediate growth suppression and inhibit the nuclear factor-kappa B pathway in pancreatic carcinoma cells. Mol Cancer Ther 2005;4:587-94.
- Foster KA, Galeffi F, Gerich FJ, Turner DA, Muller M. Optical and pharmacological tools to investigate the role of mitochondria during oxidative stress and neurodegeneration. Prog Neurobiol 2006;79:136-71.
- Lovric J, Cho SJ, Winnik FM, Maysinqeret D. Unmodified cadmium telluride quantum dots induce reactive oxygen species formation leading to multiple organelle damage and cell death. Chem Biol 2005;12:1227-34.
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for bcl-2 regulation of apoptosis. Science 1997;275:1132-6.
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, et al. Bcl-2, Bcl-xl sequester BH3 domain-only molecules preventing Bax- and Bak-mediated mitochondrial apoptosis. Mol Cell 2001;8:705-11.