
Inhibitory effect of 1,8-cineol (eucalyptol) on Egr-1 expression in lipopolysaccharide-stimulated THP-1 cells1
Introduction
Airway inflammation plays a central role in the pathogenesis of a number of lung diseases, including asthma, chronic bronchitis, bronchiectasis, and chronic obstructive pulmonary disease (COPD). Terpenes are widely used in the treatment of upper and lower airway diseases, such as chronic sinusitis and bronchitis[1,2].
As the active agent and main component of eucalyptus oil, 1,8-cineol shows particular anti-inflammation properties. Juergens et al have reported that the monoterpene 1.8-cineol revealed a steroid-like suppression of arachidonic acid metabolism, TNF-α, and interleukin (IL)-1β production in human blood monocytes in vitro[1]. Furthermore, a non-controlled study showed significant inhibition of LTB4 and IL-1β in stimulated monocytes ex vivo after additional therapy with 200 mg 1,8-cineol tid administered in enteric-coated capsules[3]. They revealed that long-term systemic therapy with 1,8-cineol had a significant steroid-saving effect in steroid-dependent asthma[2]. 1,8-Cineol significantly inhibited cytokine production in human unselected lymphocytes of TNF-α, IL-1β, IL-4, IL-5, and in lipopolysaccharide (LPS)-stimulated monocytes of TNF-α, IL-1β, IL-6, and IL-8[4]. However, studies on the mode of the anti-inflammation properties of 1,8-cineol (eucalyptol) are still infrequent.
To elucidate the anti-inflammation mechanisms of 1,8-cineol, in the present study, we investigated the effects of 1,8-cineol on the subcellular localization of early growth response factor-1 (Egr-1), the expression of Egr-1, and NF-κB in the human monocyte THP-1 cell line.
Materials and methods
Drugs and reagents 1,8-cineol was obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). LPS (from Escherichia coli 026:B6), N-α-tosyl-L-lysine chloromethyl ketone (TLCK), phorbol 12-myristate 13-acetate (PMA), protease inhibitors, and propidium iodide (PI) were purchased from Sigma (St Louis, MO, USA). 2'-Amino-3'-methoxyflavone (PD-98059) was obtained from Calbiochem (La Jolla, CA, USA). Rabbit polyclonal anti-Egr-1 and anti-NF-κB/p65 antibodies (Ab) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG, fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG were obtained from Jackson Immuno Research (West Grove, PA, USA). The gel shift assay system was from Bio-Rad (Hercules, CA, USA).
Cell culture and treatments The human monocyte cell line THP-1 was obtained from American Type Culture Collection (ATCC, N
Immunofluorescent staining Immunofluorescent staining was the same as the protocol of Yoo et al[6]. The cell monolayers, which adhered to coverslips induced by 20 µg/L PMA, were washed twice with cold PBS, then fixed with freshly prepared 3% paraformaldehyde for 15 min and permeabilized with 0.5% Triton X-100 for 15 min. After 1 h of blocking with 10% normal goat serum/PBS, the cells were incubated with the primary antibody against Egr-1 at a dilution of 1:50 in PBS for 2 h at 37 °C under a humidified atmosphere. The coverslips were washed 5 times with PBS and then incubated with FITC-conjugated IgG (1:100) diluted in PBS for 1 h at 37 °C. In order to identify the nuclei, the FITC-labeled samples were counterstained with 25 mg/L PI for 2 min.
To acquire dual-color images, the cells were examined by a 510 confocal laser scan microscope (Carl Zeiss, Oberko-chen, Germany), which was equipped with a Zeiss inverted research biological microscope and a 100×oil immersion objective (NA 1.30). The samples labeled with both FITC and PI were excited at 488 nm, and the fluorescence emissions were captured through a 510–550 nm (530 nm in center) and 590– 620 nm (605 nm in center) band pass with spectral grating, respectively.
Preparation of nuclear protein and whole cell protein extracts The nuclear protein[7] and whole cell protein extracts[8] were prepared as described with some modifica-tions. For the nuclear protein extract, after the established time of culture, the cells were collected and washed twice with cold PBS, lysed in 400 µL cold buffer A [10 mmol/L HEPES (pH 7.9), 10 mmol/L KCl, 0.1 mmol/L EDTA, and EGTA,1 mmol/L phenylmethanesulphonylfluoride (PMSF), and dithiothreitol (DTT), 1 mg/L aprotinin, leupeptin, and pepstatin A]. After 15 min, 0.1% NP-40 was added to the homogenates and the tubes were vigorously shaken for 1 min. Then the homogenates were centrifuged at 14 000 r/min at 4 °C for 5 min. The supernatant fluid (cytoplasmic extracts) was removed. The nuclear pellets were washed once with cold buffer A, then suspended in 50 µL cold buffer B [20 mmol/L HEPES (pH 7.9), 420 mmol/L NaCl, 0.1 mmol/L EDTA, and EGTA, 1 mmol/L PMSF, and DTT, and 1 mg/L aprotinin, leupeptin, and pepstatin A], and vigorously shaken at maximum speed at 4 °C for 30 min. The solution was clarified by centrifugation at 14 000 r/min for 5 min, and the supernatant fluid (nuclear extract) was stored in aliquots at -70 °C. For the whole cell protein extract, the sample cells were lysed in ice-cold buffer C [50 mmol/L Tris-HCl (pH 7.2), 150 mmol/L NaCl, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 10 mmol/L NaF, 160 µmol/L Na3VO4, and 1 mmol/L PMSF]. The suspension was cooled at 4 °C and sonicated for 2×10 s (40 W) with a probe sonicator. Following centrifugation at 14 000 r/min at 4 °C for 15 min, the supernatant fluid (whole cell protein extract) was stored in aliquots at -70 °C. The protein concentration was determined by the Folin method.
Western blot analysis The nuclear protein and whole cell protein extracts (30 µg each lane) were separated by 8% SDS-PAGE and transferred to polyvinylidene difluoride membranes using a Mini Trans-Blot module (Bio-Rad, Hercules, CA, USA). The membranes were blocked in 5% non-fat dried milk/Tris-buffered saline (TBS)/0.05% Tween 20 (blocking buffer). Then the membranes were incubated with the primary antibody in blocking buffer and washed 5 times with TBS/0.05% Tween 20 before incubation with a secondary HRP-conjugated antibody in blocking buffer (1 h, room temperature). After successive washes, the membranes were developed with an enhanced chemiluminescence kit (ECL, Santa Cruz). Anti-Egr-1 Ab and anti-NF-κB/p65 Ab, HRP-conjugated IgG were applied at a dilution of 1:2000. A semiquantitative analysis of immunoreactivity was measured by Lab Works image acquisition and analysis software (UVP GDS 8000, Upland, CA, USA), and the results were expressed as OD (optical density) value.
Statistical analysis The data are presented as mean±SEM and compared with ANOVA and least significant difference test using the SPSS statistical program(Edition 10.0, SPSS Inc, Chicago, IL, USA). The level of the statistical significance was set at P<0.05.
Results
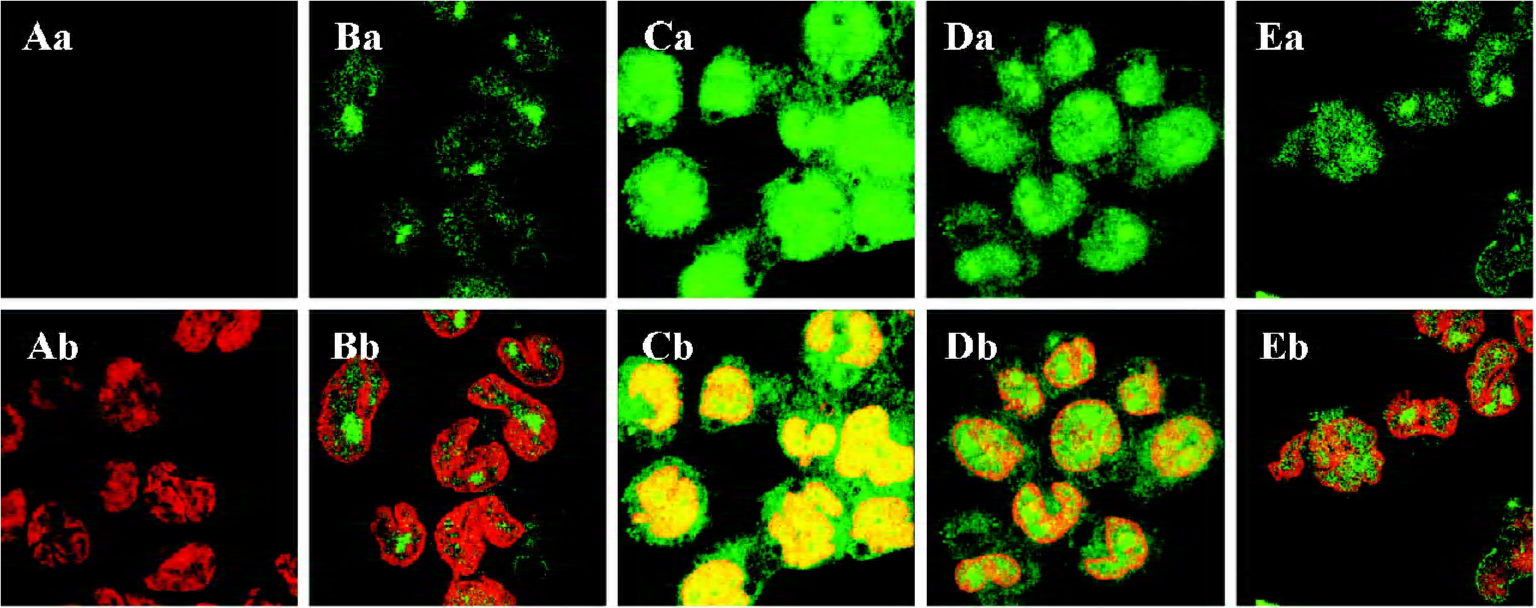
Effect of 1,8-cineol on the subcellular localization of Egr-1 in THP-1 cells The dual-color images of the FITC-labeled Egr-1 and PI-labeled nuclei in each group were detected by indirect immunofluorescence and confocal microscopy. The samples were immunocytochemically labeled with FITC for the Egr-1 protein in green, followed by incubation with the nuclear stain PI in red (Figure 1). Normal THP-1 cells were labeled in the absence of the primary antibody against Egr-1 to identify autofluorescence and nonspecific labeling. A faint or invisible signal of the FITC label (green), but only the PI label (red) was observed in these cell sheets (Figure 1A). A strong nuclear and perinuclear localization staining of Egr-1 appeared after stimulation with LPS at a concentration of 1 mg/L for 30 min in the THP-1 cells (Figure 1C) compared with the unstimulated cells (Figure 1B). The nuclear and perinuclear localization staining of Egr-1 caused by LPS was reduced by pretreatment with 100 mg/L 1,8-cineol and PD98059 (Figure 1D, 1E).
Effect of 1,8-cineol (eucalyptol) on the expression of Egr-1 in the nuclei or the whole cell The expression of Egr-1 in the nuclei and whole cell protein increased markedly after LPS stimulation (1 mg/L, 30 min). The increase induced by LPS was canceled by 1,8-cineol pretreatment in a dose-dependent manner. PD98059 dramatically inhibited LPS-induced Egr-1 expression, but TLCK did not block the induction of Egr-1 in LPS-stimulated THP-1 cells (Figures 2, 3).
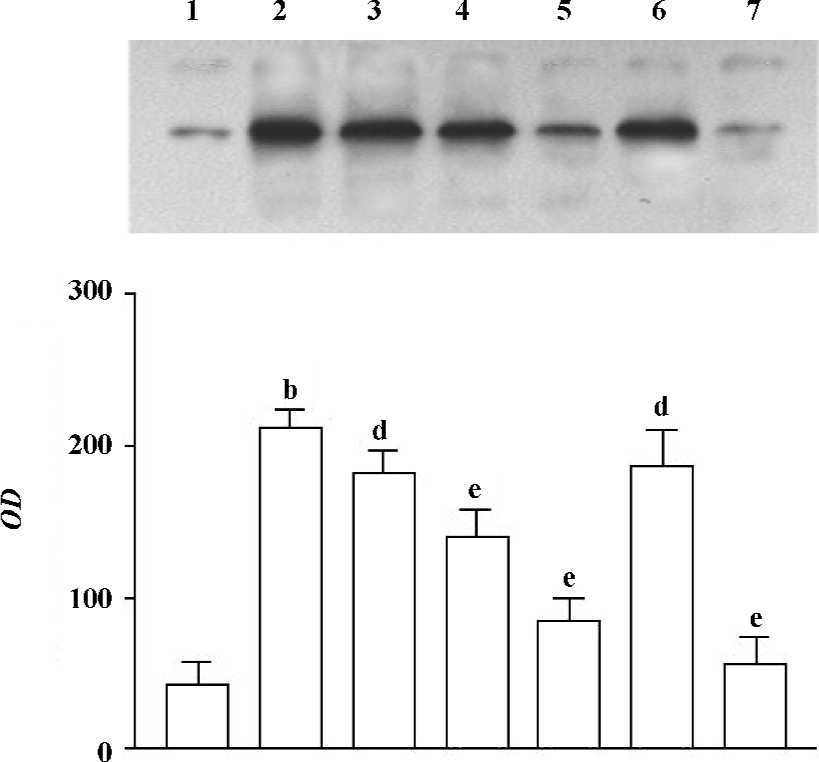
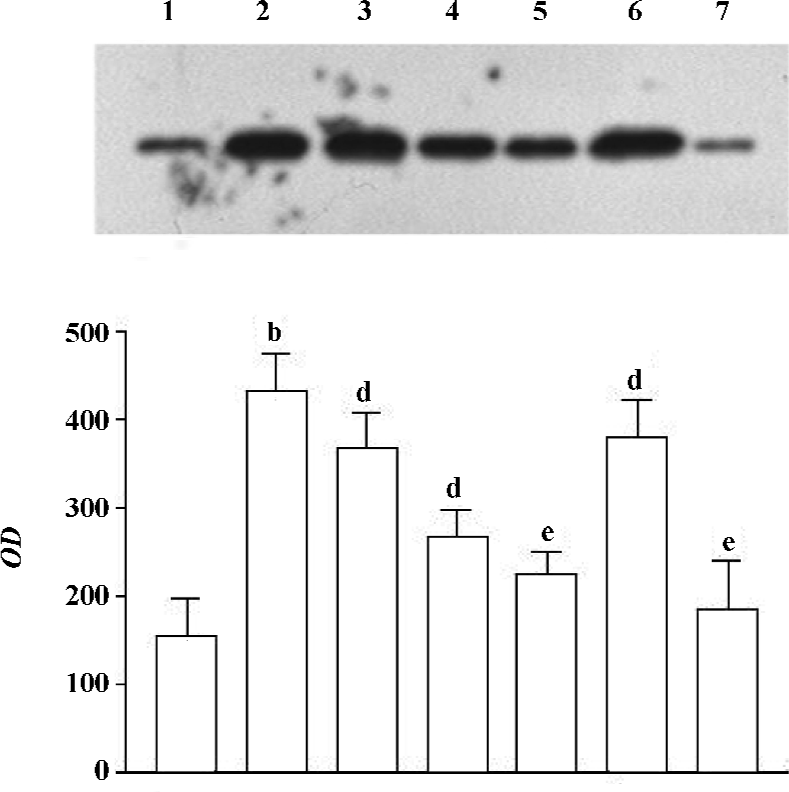
The concentration of 1,8-cineol, TLCK, and PD-98059 was determined from our previous work showing significant effects in THP-1 cells without toxicity. The cell viability, evaluated by MTT assay, did not change in the THP-1 cells at all doses used (data not show).
Effect of 1,8-cineol on the expression of NF-κB in the nuclei The NF-κB/p65 protein level in the nuclei of LPS-stimulated THP-1cells markedly increased at 30 min. There was no change on the expression of NF-κB/p65 in the nuclei after 1,8-cineol pretreatment. However, TLCK dramatically inhibited LPS induction of NF-κB/p65 expression in the nuclei (Figure 4).

Discussion
Egr-1 is a transcription factor that plays a regulatory role in the expression of many important genes for inflammation. The induction of Egr-1 has been demonstrated in cells exposed to various stimuli, including phorbol ester, ionizing radiation, inflammation, oxidative agents, and mechanical stretch/relaxation[9,10]. Its target genes include cytokine, chemokines, cell adhesion molecules, and immunoreceptors[9–12].
The human monocyte cell line THP-1 is often used as a model for tissue macrophages. Macrophages are key inflammatory cells that have been documented to play a critical role in various airway disorders[13]. Upon stimulation with molecules such as LPS, these cells secrete an array of pro-inflammatory cytokines and oxidants, including TNF-α, IL-1β, and macrophage inflammatory protein-2[14]. These pro-inflammatory cytokines genes are regulated by various transcription factors, including Egr-1 and NF-κB[15–17]. Therefore, modulation of Egr-1 or NF-κB may provide a direct way of inhibiting inflammatory mediators[18].
In this study, we investigated the effects of 1,8-cineol on the subcellular localization of Egr-1, the expression of Egr-1 in the nuclei and whole cell, and the expression of NF-κB in the nuclei of human THP-1 cells. Our studies showed that in a dose-dependent fashion, 1,8-cineol inhibited the Egr-1 synthesis and nuclear localization induced by LPS in THP-1 cells, suggesting that 1,8-cineol can inhibit the LPS-mediated Egr-1 nuclear internalization. Those results indicate that 1,8-cineol might suppress the expression of many genes important for inflammation by inhibiting Egr-1 synthesis and nuclear localization, which represents one of the anti-inflammatory mechanisms of 1,8-cineol.
In addition, PD98059, which is a selective MAP kinase kinase (MEK) inhibitor[19], dramatically inhibited LPS-induced Egr-1 expression. However, TLCK, a serine protease inhibitor[20], showed no effect on Egr-1 expression. The finding is in accordance with what Mackman et al demonstrated previously, that is that TLCK did not block the induction of Egr-1 in LPS-stimulated monocytic cells[21]. The result suggests that the inhibitory effect of 1,8-cineol on the Egr-1 expression in LPS-induced THP-1 cells primarily was due to the MEKextracellular signal-regulated kinase (ERK)1/2 pathway and not by serine protease phosphorylation pathway.
In contrast, we found that 1,8-cineol did not significantly affect the LPS-induced NF-κB expression in nuclei. As we know, Egr-1 is different from NF-κB in the signal pathway of activation. The nuclear translocation of NF-κB depends on the IκB rapid degrading through a phosphorylation-dependent and ubiquitination-dependent mechanism[22]. However, mitogenic stimulation of the Egr-1 gene has been shown to be mediated in many cell types through the RasRaf-1-MEK-ERK1/2 signal transduction pathway[19,23,24]. Thus, the anti-inflammation mechanisms of 1,8-cineol is more likely related to Egr-1 rather than NF-κB.
In conclusion, these observations suggest that 1,8-cineol might block the effect of Egr-1 by inhibiting the synthesis of Egr-1 and preventing Egr-1 nuclear internalization, which might at least represent one of the anti-inflammatory mechanisms of 1,8-cineol. This provides evidence for the role of 1,8-cineol in controlling airway inflammation.
References
- Juergens UR, Stober M, Vetter H. Inhibition of cytokine production and arachidonic acid metabolism by eucalyptol (1.8-cineole) in human blood monocytes in vitro. Eur J Med Res 1998;3:508-10.
- Juergens UR, Dethlefsen U, Steinkamp G, Gillissen A, Repges R, Vetter H. Anti-inflammatory activity of 1.8-cineol (eucalyptol) in bronchial asthma: a double-blind placebo-controlled trial. Respir Med 2003;97:250-6.
- Juergens UR, Stober M, Schmidt-Schilling L, Kleuver T, Vetter H. Antiinflammatory effects of eucalyptol (1.8-cineole) in bronchial asthma: inhibition of arachidonic acid metabolism in human blood monocytes ex vivo. Eur J Med Res 1998;3:407-12.
- Juergens UR, Engelen T, Racke K, Stober M, Gillissen A, Vetter H. Inhibitory activity of 1,8-cineol (eucalyptol) on cytokine production in cultured human lymphocytes and monocytes. Pulm Pharmacol Ther 2004;17:281-7.
- Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer 1980;26:171-6.
- Yoo CG, Lee S, Lee CT, Kim YW, Han SK, Shim YS. Effect of acetylsalicylic acid on endogenous I kappa B kinase activity in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 2001;280:L3-9.
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acid Res 1989;17:6419.
- Yin Y, Allen PD, Jia L, Kelsey SM, Newland AC. 8-Cl-adenosine mediated cytotoxicity and sensitization of T-lymphoblastic leukemia cells to TNFalpha-induced apoptosis is via inactivation of NF-kappaB. Leuk Res 2001;25:423-31.
- Liu L, Tsai JC, Aird WC. Egr-1 gene is induced by the systemic administration of the vascular endothelial growth factor and the epidermal growth factor. Blood 2000;96:1772-81.
- Kamimura M, Viedt C, Dalpke A, Rosenfeld ME, Mackman N, Cohen DM, et al. Interleukin-10 suppresses tissue factor expression in lipopolysaccharide-stimulated macrophages via inhibition of Egr-1 and a serum response element/MEK-ERK1/2 pathway. Circ Res 2005;97:305-13.
- Ke J, Gururajan M, Kumar A, Simmons A, Turcios L, Chelvarajan RL, . The role of MAP kinases in BCR induced down-regulation of Egr-1 in immature B lymphoma cells. J Biol Chem 2006; 281: 39 806–18.
- Cho SJ, Kang MJ, Homer RJ, Kang HR, Zhang X, Lee PJ, et al. Role of early growth response-1 (Egr-1) in interleukin-13-induced inflammation and remodeling. J Biol Chem 2006;281:8161-8.
- Yao PL, Tsai MF, Lin YC, Wang CH, Liao WY, Chen JJ, et al. Global expression profiling of theophylline response genes in macrophages: evidence of airway anti-inflammatory regulation. Respir Res 2005;6:89.
- Ye Q, Chen B, Tong Z, Nakamura S, Sarria R, Costabel U, et al. Thalidomide reduces IL-18, IL-8 and TNF-alpha release from alveolar macrophages in interstitial lung disease. Eur Respir J 2006;28:824-31.
- Yao J, Mackman N, Edgington TS, Fan ST. Lipopolysaccharide induction of the tumor necrosis factor-α promoter in human monocytic cells: regulation by Egr-1, c-Jun, and NF-κB transcription factors. J Biol Chem 1997;272:17795-801.
- Xu Z, Dziarski R, Wang Q, Swartz K, Sakamoto KM, Gupta D. Bacterial peptidoglycan-induced TNF-alpha transcription is mediated through the transcription factors Egr-1, Elk-1, and NF-kappaB. J Immunol 2001;167:6975-82.
- Nagel T, Resnick N, Dewey CF Jr, Gimbrone MA Jr. Vascular endothelial cells respond to spatial gradients in fluid shear stress by enhanced activation of transcription factors. Arterioscler Thromb Vasc Biol 1999;19:1825-34.
- Emery JG, Ohlstein EH, Jaye M. Therapeutic modulation of transcription factor activity. Trends Pharmacol Sci 2001;22:233-40.
- Guha M, O’Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, et al. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood 2001;98:1429-39.
- Jeong JY, Kim KU, Jue DM. Tosylphenylalanine chloromethyl ketone inhibits TNF-alpha mRNA synthesis in the presence of activated NF-kappa B in RAW 264.7 macrophages. Immunology 1997;92:267-73.
- Mackman N. Protease inhibitors block lipopolysaccharide induction of tissue factor gene expression in human monocytic cells by preventing activation of c-Rel/p65 heterodimers. J Biol Chem 1994; 269: 26 363–7.
- Baldwin AS Jr. The NF-κB and I κB proteins: new discoveries and insights. Annu Rev Immunol 1996;14:649-83.
- Schwachtgen JL, Houston P, Campbell C, Sukhatm VP, Braddock M. Fluid shear stress activation of egr-1 transcription in cultured human endothelial and epithelial cells is mediated via the extracellular signal-related kinase 1/2 mitogen-activated protein kinase pathway. J Clin Invest 1998;101:2540-9.
- Dieckgraefe BK, Weems DM. Epithelial injury induces egr-1 and fos expression by a pathway involving protein kinase C and ERK. Am J Physiol 1999;276:G322-30.