
Mitochondrial-derived ROS in edelfosine-induced apoptosis in yeasts and tumor cells1
Introduction
The anticancer drug 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine (ET-18-OCH3, edelfosine), is the prototypic compound of a promising family of antitumor drugs, collectively known as alkyl-lysophospholipid analogs or antitumor ether lipids, which induce apoptosis in a wide variety of cancer cells[1-4]. Edelfosine has been also found to be cytotoxic to Saccharomyces cerevisiae yeasts[5,6], but the type of cell death induced by edelfosine in yeasts remains to be established. Recent evidence suggests that edelfosine-induced cytotoxicity in both tumor cells and yeasts is mediated through a modification of lipid raft composition[6-8]. In addition, mitochondria[9,10] and endoplasmic reticulum[11] have been suggested to play a major role in edelfosine-mediated apoptosis in tumor cells.
Mitochondria have a central role in apoptosis, and many important aspects of the apoptotic process converge in this organelle[12]. Mitochondria are the richest source of reactive oxygen species (ROS) in the cell, converting 1%-2% of reduced oxygen into superoxide anion[13]. The inhibition of the mitochondrial electron transport chain, resulting in subsequent release of ROS, is an early event in apoptotic cell death induced by different agents (eg, ceramide, dexametha-sone, and bleomycin)[14-16].
Although it was initially assumed that apoptosis arose with multicellularity and would have been counterselected in unicellular organisms, several findings indicate that a similar process operates in single-celled eukaryotes[17,18]. In particular, the yeast S cerevisiae has been reported to undergo apoptosis-like cell death in response to various harsh treatments[19].
In the present report, we have found that edelfosine induces apoptosis in S cerevisiae cells via mitochondrial-derived ROS generation. In addition, edelfosine-induced apoptosis in human tumor cells was also mediated by mitochondria and correlated with ROS generation, suggesting that a similar process mediates cytotoxicity of this drug in both yeasts and human tumor cells.
Materials and methods
Cell culture and reagents The S cerevisiae strain used in this study was BY4742 (MATα his3Δ leu2Δ lys2Δ ura3Δ). The cells were incubated at 32 °C in a 25-mL flask with 10 mL YPAD (1% yeast extract, 2% peptone, 2% D-glucose, and 0.024 mg/ml adenine hemisulfate) medium with constant shaking at 200 ×g. Cell growth was monitored by measuring the optical density of the cultures at 600 nm (OD600) by using an spectrophotometer. After 2 d incubation at 32 °C, the yeasts were at the stationary phase (OD600= 6–10) and then cells were diluted for the experiments.
Human acute T-cell leukemia Jurkat and CEM-C7H2 cells were cultured at 37 °C in a humidified atmosphere of 5% CO2 in RPMI-1640 medium supplemented with 10% (v/v) heat-inactivated fetal calf serum (FCS), 2 mmol/L L-gluta-mine, 100 U/mL penicillin, and 100 µg/mL strepto-mycin. The CEM-C7H2-9F3 cells, stably transfected with human transgenic Bcl-2[20], were kindly provided by Dr S Geley (Institute for General and Experimental Pathology, University of Innsbruck, Innsbruck, Austria) and cultured in the presence of 500 μg/mL hygromycin as previously described[9].
Edelfosine was obtained from Huabei Biochem (Beijing, China) and INKEYSA (Barcelona, Spain). Stock solutions of edelfosine in sterile water (for yeast studies) and RPMI-1640 culture medium containing 10% heat-inactivated FCS (for Jurkat cell studies) were prepared as previously described[9]. All other chemicals were from Sigma (St Louis, MO, USA).
Edelfosine treatment of S cerevisiae The yeasts were inoculated into a 25-mL flask containing 10 mL YPAD medium. Starter yeast cultures were grown to saturation (about 2 d) at 32 °C (approximately 1×108 cells/mL). After saturation, cells were diluted with fresh YPAD medium to an OD600=0.1. Then, 10 mL of this diluted cell suspension was added to pre-warmed 25 mL Erlenmeyer flasks containing 10 mL YPAD medium, and incubated with increasing concentrations of edelfosine at 32 °C, with shaking at 200 ×g. Then, 1 mL of cultured cells were taken at 2-h intervals to determine cell proliferation by absorbance readings at OD600 and to measure the generation of ROS. Furthermore, to identify whether ROS generation is the trigger of apoptosis, the antioxidant α-tocopherol succinate (Sigma, USA) was used with edelfosine. α-Tocopherol succinate was dissolved in sterile absolute ethanol at a concentration of 10 mmol/L and stored at -20 °C. For the cell treatment, α-tocopherol was added to the cell culture medium to achieve a final concentration of 10 μmol/L.
ROS measurement in yeasts A modified version of a previously described assay for the intracellular conversion of nitro blue tetrazolium (NBT) to formazan by superoxide anion was used to measure the generation of ROS[21]. Briefly, the yeasts were incubated with 50 µmol/L NBT for 15 min at 32 °C, fixed in absolute ethanol, and air dried. The formazan content of the cells was then solubilized with 960 µL 2 mol/L potassium hydroxide and 1120 µL DMSO. The absorbance at 630 nm was measured spectrophoto-metrically. As a positive control, 100 µmol/L H2O2 was added to the cells and the amount of formazan formed was measured.
Inhibition of mitochondrial function Rotenone, an inhibitor of complex I of the mitochondria electron transport chain[22], was dissolved in tissue culture grade DMSO to achieve a final concentration of 1 mmol/L. For the cell treat-ments, rotenone was added at a final concentration of 50 nmol/L to the cell culture, and incubated at 32 °C, with constant shaking at 200 ×g for 6 h. The solvent concentration in the media never exceeded 0.001%.
Assessment of apoptosis by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay The cells were loaded on coverslides coated with poly-L-lysine (Sigma, USA) and dried at 37 °C. After incubation for 5 min, the cells were fixed in 4% paraformaldehyde for 20 min at 4 °C. The cells were then gently washed twice with phosphate buffered saline (PBS) and permeabilized with 0.2% Triton X-100 for 5 min. Following 2 PBS washes, TUNEL assay was carried out with the Promega kit (DeadEnd Fluorometric TUNEL System, Promega, Madison, WI, USA) according to the manufacturer抯 instructions. Finally, the cells were analyzed immediately using a Leica DMRXA microscope (Leica, Wetzlar, Germany) equipped with Nomarski optics and epifluorescence and photographed with a Photometrics Sensys CCD camera (Tucson, AZ, USA). The percentage of cells undergoing apoptosis was determined by counting the number of cells with apoptotic staining in 10 randomly selected microscope fields (10× ocular, 40× objective).
Assay of mitochondrial membrane potential In order to explore whether edelfosine changes mitochondrial membrane potential in yeast and human acute T-cell leukemia Jurkat cells, the cells were incubated with 2 mmol/L rhodamine123 for 10 min at 37 °C. After the incorporation of the fluorescent probe, the cells were incubated with or without 5 μmol/L edelfosine and treated for 6 h. At the end of the incubation, the cells were washed twice with PBS, and then resuspended in 1.5 mL PBS. For the yeast cells, the samples were sonicated for 5 s at 8 microns just before passing through the flow cytometer. The fluorescence of each individual cell was measured with a flow cytometer at an excitation wavelength of 480 nm and an emission wavelength of 530 nm, and analyzed with CellQuest 3.1f Analysis Software (Becton Dickinson, Franklin Lakes, NJ, USA).
Cytofluorimetric analysis of DNA fragmentation and generation of ROS in Jurkat cells The quantitation of apoptotic cells was calculated by flow cytometry as the percentage of cells in the sub-G1 region (hypodiploidy) in cell cycle analysis as previously described[23]. To evaluate the generation of ROS, the cells were incubated in PBS with 2 μmol/L dihydroethidine (HE) for 20 min at 37 ºC, followed by analysis on a Becton-Dickinson FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA) flow cytometer as previously described[9].
Statistical analysis Statistical evaluation of the effect of edelfosine was performed by analysis of variance (ANOVA). Statistical significance was recognized at P<0.05.
Results
Edelfosine induces apoptosis in S cerevisiae We found that edelfosine inhibited yeast proliferation in a concentration-dependent manner (Figure 1A). This effect was cytotoxic rather than cystostatic as cell viability declined with increasing concentrations of edelfosine when cultured again in drug-free liquid medium (data not shown). We next determined whether edelfosine-induced yeast death by apoptosis. One of the most important markers of apoptosis is chromosomal DNA fragmentation as assessed by TUNEL staining, a reliable marker of yeast apoptosis[24,25]. Propidium iodide stains both apoptotic and non-apoptotic cells, but fluorescein-12-dUTP incorporation results in localized green fluorescence within the nuclei of only the DNA-degraded apoptotic cells. Most of the cells treated with either 2.5 or 5 µmol/L edelfosine for 6 h were TUNEL positive, whereas all of the untreated cells remained TUNEL negative (Figure 1B). These results indicate that edelfosine-induced cell death in yeasts by apoptosis as previously shown in tumor cells[1,2,9].
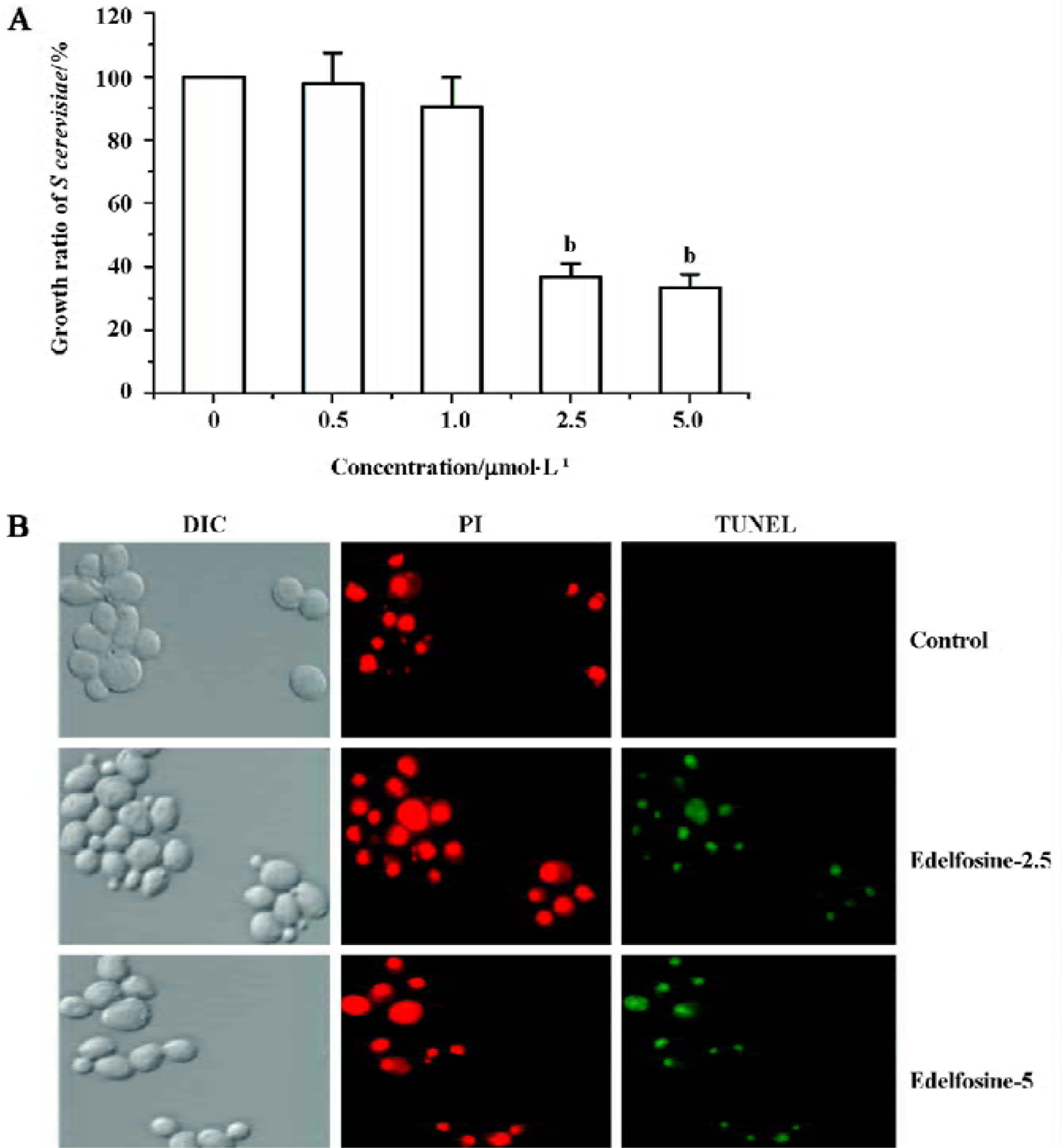
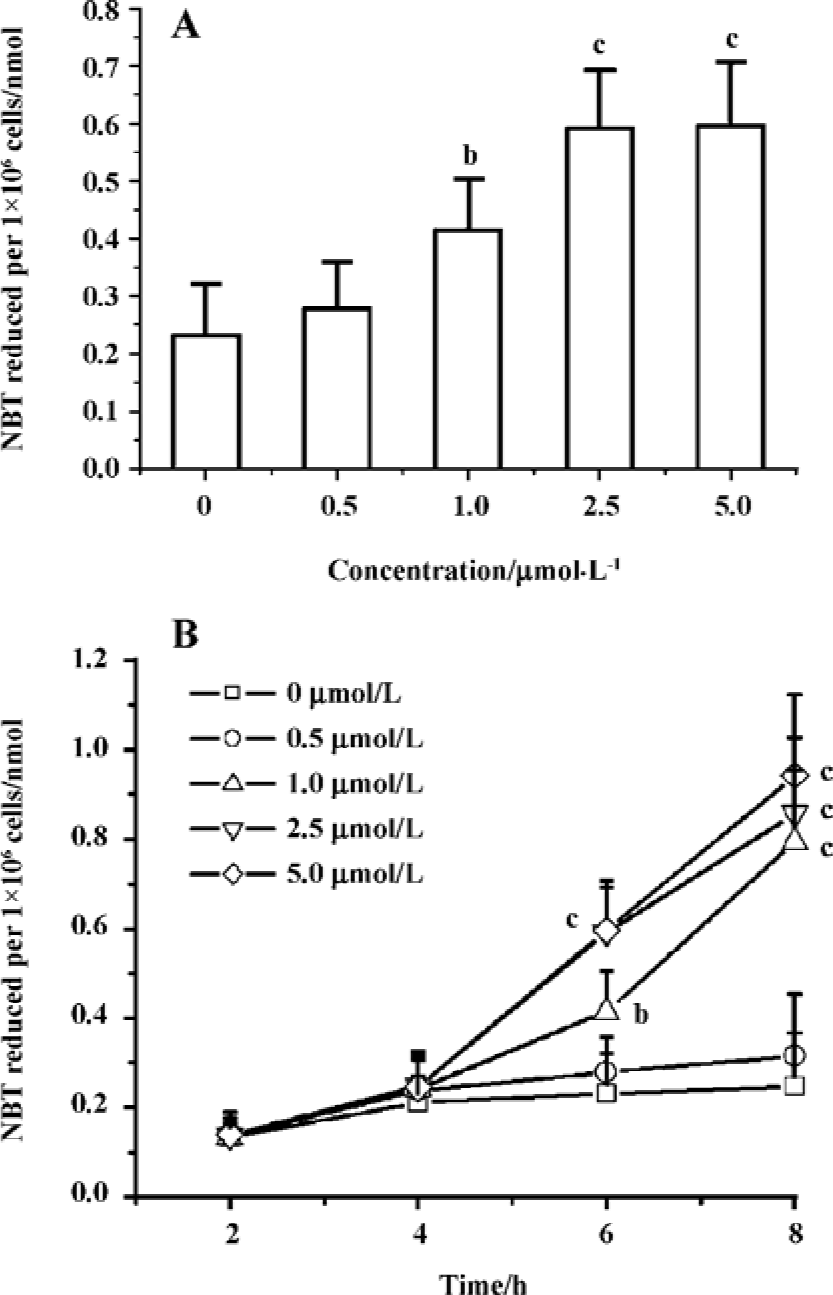
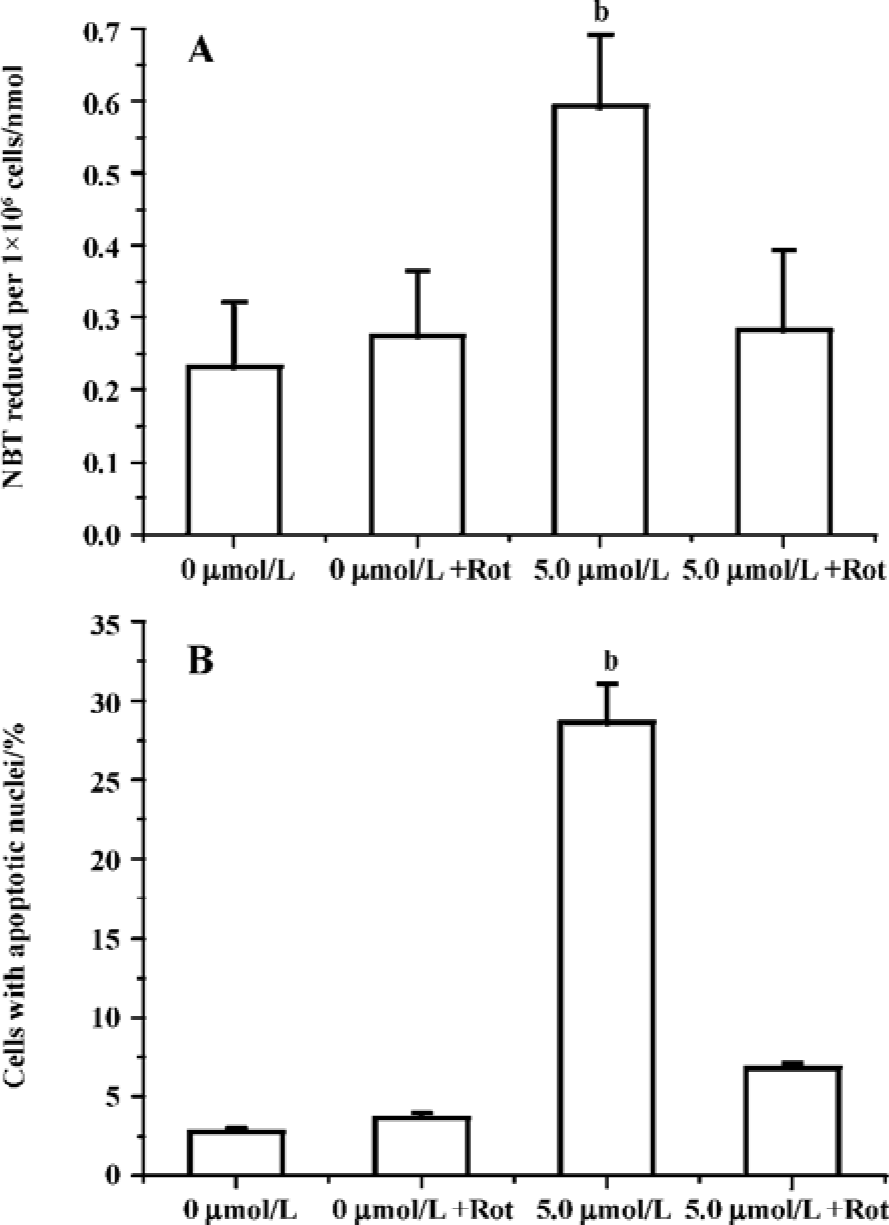
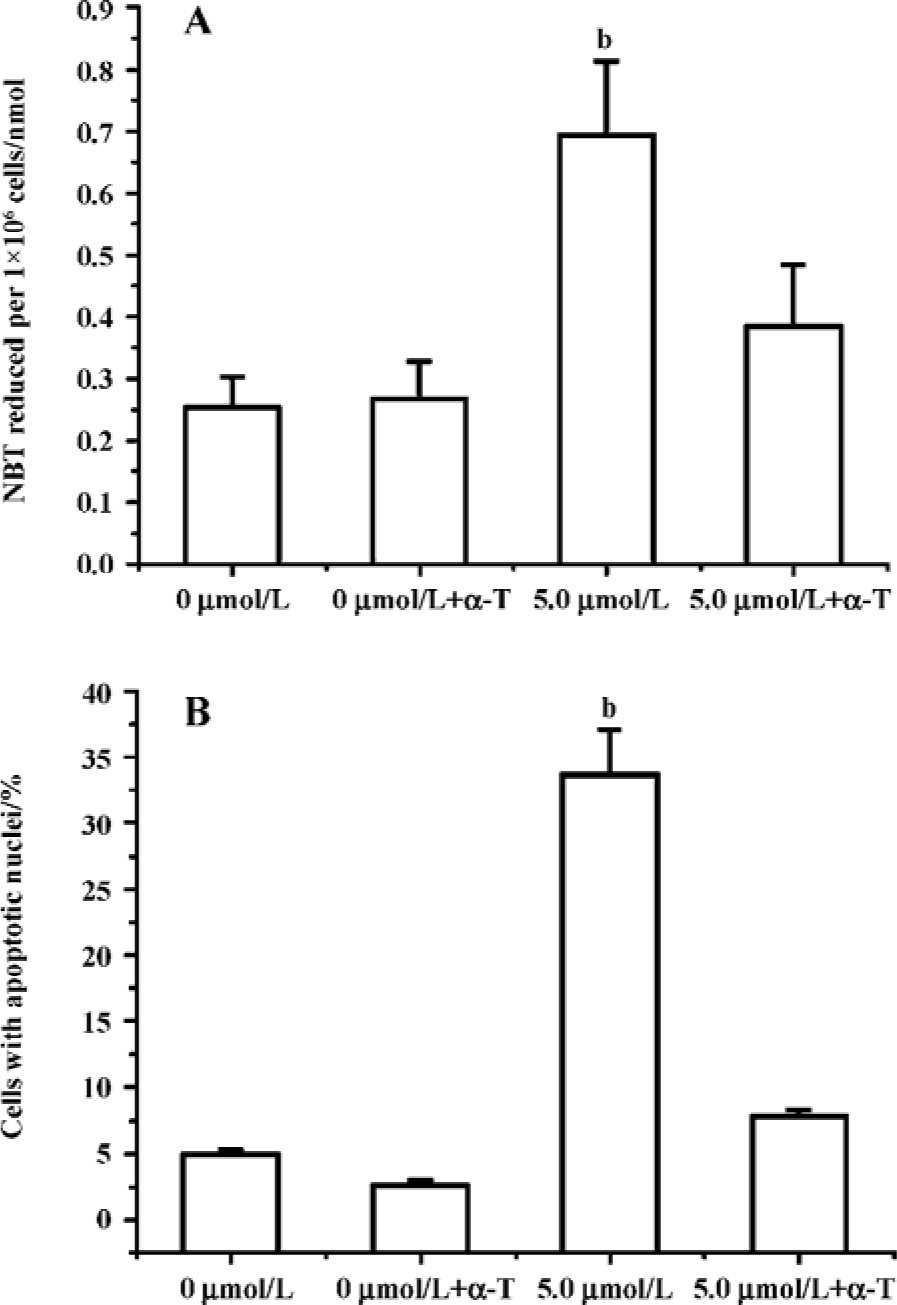
Edelfosine induces apoptosis in S cerevisiae via mitochondrial-derived ROS The external addition of H2O2 has been reported as a major inducer of apoptosis in yeasts[24]. Yeast strain YPH98gsh1, lacking glutathione due to a deletion of the GSH1 gene coding γ-glutamylcysteine synthetase, undergoes apoptosis after 3 d of being cultured with glutathione-free medium[24,25]. Thus, ROS generation seems a key event in yeast apoptosis. We next analyzed the putative increase in ROS following yeast treatment with edelfosine. We found that the alkyl-lysophospholipid analog induced a concentration- and time-dependent increase in the generation of ROS. The results showed that the generation of ROS was significantly increased in S cerevisiae cells treated with 1.0, 2.5, and 5.0 µmol/L edelfosine for 6 and 8 h as compared to the control (Figure 2A,B). The addition of rotenone, an inhibitor of the complex I in the mitochondrial electron chain, prior to edelfosine treatment prevented both ROS generation (Figure 3A) and apoptosis (Figure 3B) in S cerevisiae cells. Edelfosine-induced generation of intracellular ROS and apoptosis were also abrogated by 10 µmol/L α-tocopherol (Figure 4). These data suggest that ROS are generated from mitochondria upon edelfosine treatment in yeasts and that ROS generation was the trigger of apoptosis.
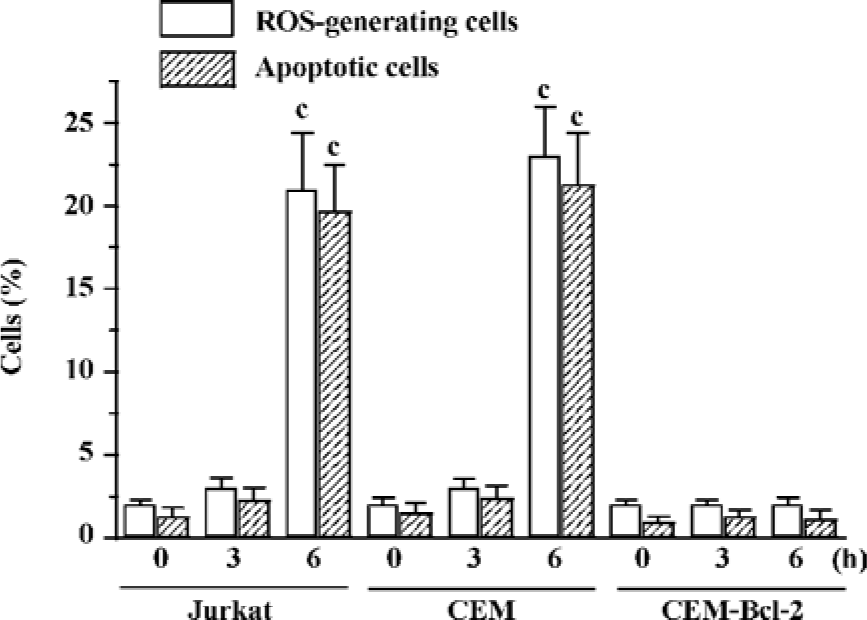
Edelfosine induces mitochondrial-derived ROS in human tumor cells The above data indicated that ROS generated in mitochondria played a role in edelfosine-induced apoptosis in yeasts. We next studied whether the same process took place in human tumor cells. We found that edelfosine induced ROS generation in human leukemia Jurkat and CEM-C7H2 cells that preceded apoptosis (Figure 5). Since Bcl-2 acts as a safeguard in mitochondria[12], we analyzed the effect of overexpressing Bcl-2 in edelfosine-induced apoptosis and ROS generation in CEM-C7H2 cells. The ectopic expression of Bcl-2 blocked both ROS generation and apoptosis when the cells were incubated with edelfosine (Figure 5), suggesting that mitochondria and mitochondrial-derived ROS generation may play a role in edelfosine-induced apoptosis in human tumor cells.
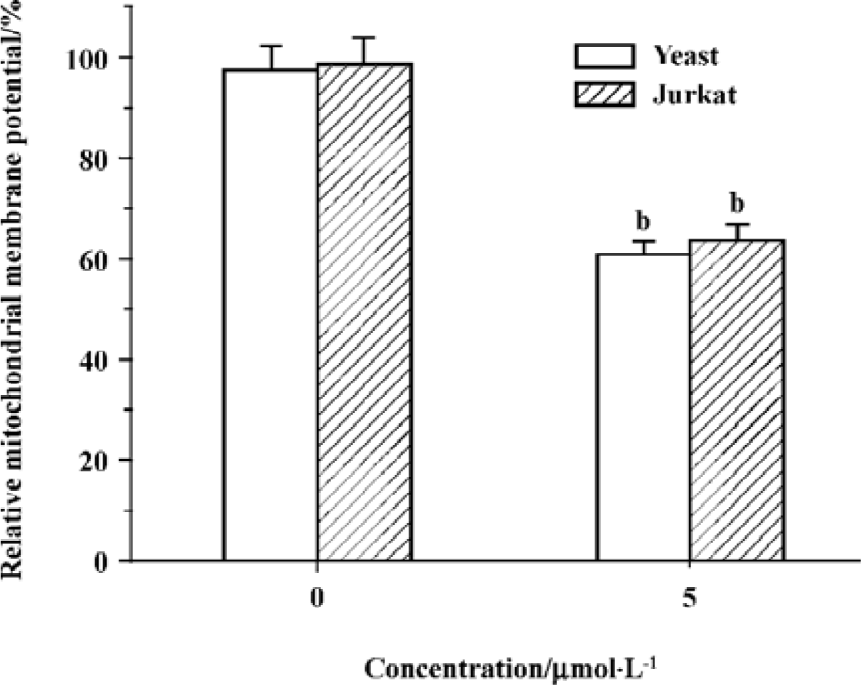
Edelfosine induces changes of mitochondrial membrane potential In the present study, the results showed that relative mitochondrial membrane potential in yeast and Jurkat cell treated with 5 祄ol/L edelfosine for 6 h decreased significantly (Figure 6), suggesting functional alterations in mitochondria.
Discussion
Edelfosine has been reported to be preferentially accumulated in the plasma membrane of transformed cells, accounting for up to 17% of the purified membrane phospho-lipids. Despite edelfosine exerts its major effects at the plasma membrane level, it also affects gene expression by modulating the expression and activity of transcription factors[3,4]. Current evidence indicates that edelfosine induces apoptosis through mitochondria. Edelfosine-induced apoptosis is prevented by Bcl-2 or Bcl-xL overexpression by gene transfer[2]. Edelfosine induces the disruption of the mitochondrial transmembrane potential followed by the production of ROS and the release of mitochondrial cytochrome c[9]. In addition, edelfosine activates caspase-3 and its inactivation prevents the apoptotic response elicited by the ether lipid[9]. Moreover, recent evidence indicates that edelfosine is the first antitumor drug acting through the intracellular activation of the Fas/CD95 death receptor, involving a raft-mediated pro-cess[7,8].
We have described here that the antitumor agent edelfosine induces apoptosis in yeasts through a mitochondrial-generated ROS process. It reacts similarly in human tumor cells, which suggests that this mechanism is an ancestral apoptotic pathway being preserved in both single-celled and metazoan organisms. S cerevisiae has become a powerful model organism with which to study the process of apoptosis[26,27]. We propose that it may contribute to exploring the molecular mechanism of edelfosine on apoptosis. Moreover, some of its key attributes and advantages in this regard are the ease with which genetic, molecular, and cytological manipulations can be performed in this yeast.
Our results indicate that yeasts undergo apoptosis when treated with edelfosine. DNA fragmentation in S cerevisiae, for the determination of apoptosis, has so far only been evidenced by TUNEL-positive phenotypes[24,25]. This is apparently due to the induction of large DNA fragments (several hundreds kilobases) in yeast apoptosis[25], rather than the typical 180 bp multiple DNA fragments found in mammalian cell apoptosis following internucleosomal DNA fragmentation[28]. The TUNEL assay was developed for measuring DNA fragmentation at the single-cell level by incorporating biotylated or fluorescent dUTP at sites of free 3' -hydroxy groups in DNA[29]. The TUNEL assay has been used in numerous studies to classify apoptotic cell death in yeasts[24,25]. Our results showed that a majority of yeasts treated with 2.5 or 5 祄ol/L edelfosine were TUNEL-positive, suggesting apoptosis in the yeast cell.
Apoptosis is characterized by biochemical processes that are largely conserved throughout evolution, and mitochondria seem to act as a central coordinator of cell death[30,31]. A number of reports have revealed that perturbations in mitochondria respiration occur early in the apoptotic process and that the mitochondrion itself serves as a control switch for apoptosis[32,33]. Complex I (NADH-quinone oxido-reductase) of the mitochondrial electron transport chain has been reported to be an important site for leakage of electrons and the subsequent generation of ROS. Complex I contains a single large inhibitor binding pocket[34], and binding to this site of different types of inhibitors, including rotenone[35], prevents mitochondrial ROS production[14,16]. In this study, rotenone prevented both apoptosis and ROS generation in yeasts following edelfosine treat-ment. The results showed that edelfosine induces the disruption of the mitochondrial transmembrane potential followed by the production of ROS that later could lead to the onset of apoptosis.
Our data also showed that edelfosine increased ROS in human leukemic cells and the overexpression of Bcl-2 blocked both apoptosis and ROS generation, in agreement with previous findings[9]. Thus, a similar mitochondrial-mediated ROS production is taking place in edelfosine-promoted cell death in both yeasts and human tumor cells, suggesting that yeasts might be used as a model to study the mechanism of action of edelfosine. Our data also support a major role for mitochondria and mitochondrial-derived ROS in edelfosine-induced cell death.
Collectively, our findings have shown for the first time that the antitumor agent edelfosine induces an apoptotic response in yeasts that involves mitochondria, most likely through the generation of mitochondrial-derived ROS. This deadly process induced by edelfosine in yeasts is similar to the cell death process triggered by this drug in human tumor cells, suggesting that this mechanism is an ancestral apoptotic pathway being preserved in both single-celled and metazoan organisms. Interestingly, these results suggest that S cerevisiae might be used as an interesting cell model to study the mechanism of action of edelfosine.
References
- Mollinedo F, Martinez-Dalmau R, Modolell M. Early and selective induction of apoptosis in human leukemic cells by the alkyl-lysophospholipid ET-18-OCH3. Biochem Biophys Res Commun 1993;192:603-9.
- Mollinedo F, Fernandez-Luna JL, Gajate C, Martin-Martin B, Benito A, Martinez-Dalmau R, et al. Selective induction of apoptosis in cancer cells by the ether lipid ET-18-OCH3 (Edelfo-sine): molecular structure requirements, cellular uptake, and protection by Bcl-2 and Bcl-XL. Cancer Res 1997;57:1320-8.
- Gajate C, Mollinedo F. Biological activities, mechanisms of action and biomedical prospect of the antitumor ether phospholipid ET-18-OCH3 (edelfosine), a proapoptotic agent in tumor cells. Curr Drug Metab 2002;3:491-525.
- Mollinedo F, Gajate C, Martin-Santamaria S, Gago F. ET-18-OCH3 (edelfosine): a selective antitumour lipid targeting apoptosis through intracellular activation of Fas/CD95 death receptor. Curr Med Chem 2004;11:3163-84.
- Hanson PK, Malone L, Birchmore JL, Nichols JW. Lem3p is essential for the uptake and potency of alkylphosphocholine drugs, edelfosine and miltefosine. J Biol Chem 2003;278:36041-50.
- Zaremberg V, Gajate C, Cacharro LM, Mollinedo F, McMaster CR. Cytotoxicity of an anti-cancer lysophospholipid through selective modification of lipid raft composition. J Biol Chem 2005;280:38047-58.
- Gajate C, Mollinedo F. The antitumor ether lipid ET-18-OCH3 induces apoptosis through translocation and capping of Fas/CD95 into membrane rafts in human leukemic cells. Blood 2001;98:3860-3.
- Gajate C, Del Canto-Janez E, Acuna AU, Amat-Guerri F, Geijo E, Santos-Beneit AM, et al. Intracellular triggering of Fas aggregation and recruitment of apoptotic molecules into Fas-enriched rafts in selective tumor cell apoptosis. J Exp Med 2004;200:353-65.
- Gajate C, Santos-Beneit AM, Macho A, Lazaro M, Hernandez-De Rojas A, Modolell M, et al. Involvement of mitochondria and caspase-3 in ET-18-OCH3-induced apoptosis of human leukemic cells. Int J Cancer 2000;86:208-18.
- Vrablic AS, Albright CD, Craciunescu CN, Salganik RI, Zeisel SH. Altered mitochondrial function and overgeneration of reactive oxygen species precede the induction of apoptosis by 1-O-octadecyl-2-methyl-rac-glycero-3-phosphocholine in p53-defective hepatocytes. FASEB J 2001; 15: 1739-44.
- Nieto-Miguel T, Gajate C, Mollinedo F. Differential targets and subcellular localization of antitumor alkyl-lysophospholipid in leukemic versus solid tumor cells. J Biol Chem 2006;281:14833-40.
- Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309-12.
- Richter C, Gogvadze V, Laffranchi R, Schlapbach R, Schweizer M, Suter M, et al. Oxidants in mitochondria: from physiology to diseases. Biochim Biophys Acta 1995;1271:67-74.
- Castedo M, Macho A, Zamzami N, Hirsch T, Marchetti P, Uriel J, et al. Mitochondrial perturbations define lymphocytes undergoing apoptotic depletion in vivo. Eur J Immunol 1995;25:3277-84.
- Hug H, Strand S, Grambihler A, Galle J, Hack V, Stremmel W, et al. Reactive oxygen intermediates are involved in the induction of CD95 ligand mRNA expression by cytostatic drugs in hepatoma cells. J Biol Chem 1997;272:28191-3.
- Quillet-Mary A, Jaffrezou JP, Mansat V, Bordier C, Naval J, Laurent G. Implication of mitochondrial hydrogen peroxide generation in ceramide-induced apoptosis. J Biol Chem 1997;272:21388-95.
- Shaha C. Apoptosis in Leishmania species and its relevance to disease pathogenesis. Indian J Med Res 2006;123:233-44.
- Madeo F, Engelhardt S, Herker E, Lehmann N, Maldener C, Proksch A, et al. Apoptosis in yeast: a new model system with applications in cell biology and medicine. Curr Genet 2002;41:208-16.
- Phillips AJ, Sudbery I, Ramsdale M. Apoptosis induced by environmental stresses and amphotericin B in Candida albicans. Proc Natl Acad Sci USA 2003;100:14327-32.
- Geley S, Hartmann BL, Kofler R. Ceramides induce a form of apoptosis in human acute lymphoblastic leukemia cells that is inhibited by Bcl-2, but not by CrmA. FEBS Lett 1997;400:15-8.
- Rook GA, Steele J, Umar S, Dockrell HM. A simple method for the solubilisation of reduced NBT, and its use as a colorimetric assay for activation of human macrophages by gamma-interferon. J Immunol Methods 1985;82:161-7.
- Lopez-Barneo J, Ortega-Saenz P, Piruat JI, Garcia-Fernandez M. Oxygen-sensing by ion channels and mitochondrial function in carotid body glomus cells. Novartis Found Symp 2006;272:54-64.
- Gajate C, Barasoain I, Andreu JM, Mollinedo F. Induction of apoptosis in leukemic cells by the reversible microtubule-disrupting agent 2-methoxy-5-(2',3',4'-trimethoxyphenyl)-2,4,6-cycloheptatrien-1-one: protection by Bcl-2 and Bcl-XL and cell cycle arrest. Cancer Res 2000;60:2651-9.
- Madeo F, Frohlich E, Ligr M, Grey M, Sigrist SJ, Wolf DH, et al. Oxygen stress: a regulator of apoptosis in yeast. J Cell Biol 1999;145:757-67.
- Ribeiro GF, Corte-Real M, Johansson B. Characterization of DNA damage in yeast apoptosis induced by hydrogen peroxide, acetic acid, and hyperosmotic shock. Mol Biol Cell 2006;17:4584-91.
- Herker E, Jungwirth H, Lehmann KA, Maldener C, Frohlich KU, Wissing S, et al. Chronological aging leads to apoptosis in yeast. J Cell Biol 2004;164:501-7.
- Fabrizio P, Battistella L, Vardavas R, Gattazzo C, Liou LL, Diaspro A, et al. Superoxide is a mediator of an altruistic aging program in Saccharomyces cerevisiae. J Cell Biol 2004;166:1055-67.
- Mollinedo F, Gajate C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003;8:413-50.
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 1992;119:493-501.
- Gottlieb RA. Mitochondrial signaling in apoptosis: mitochondrial daggers to the breaking heart. Basic Res Cardiol 2003;98:242-9.
- Gottlieb RA. Role of mitochondria in apoptosis. Crit Rev Eukaryot Gene Exp 2000;10:231-9.
- Decaudin D, Marzo I, Brenner C, Kroemer G. Mitochondria in chemotherapy-induced apoptosis: a prospective novel target of cancer therapy. Int J Oncol 1998;12:141-52.
- Pinkoski MJ, Waterhouse NJ, Green DR. Mitochondria, apoptosis and autoimmunity. Curr Dir Autoimmun 2006;9:55-73.
- Okun JG, Zickermann V, Zwicker K, Schagger H, Brandt U. Binding of detergents and inhibitors to bovine complex I - a novel purification procedure for bovine complex I retaining full inhibitor sensitivity. Biochim Biophys Acta 2000;1459:77-87.
- Schuler F, Yano T, Di Bernardo S, Yagi T, Yankovskaya V, Singer TP, et al. NADH-quinone oxidoreductase: PSST subunit couples electron transfer from iron-sulfur cluster N2 to quinone. Proc Natl Acad Sci USA 1999;96:4149-53.