
Upregulation of PTEN involved in rosiglitazone-induced apoptosis in human hepatocellular carcinoma cells1
Introduction
Hepatocellular carcinoma (HCC) affects more than 500 000 people worldwide annually, and the 5-year mortality exceeds 95%. More than half of those people are in China[1]. Present measures, including mainly surgical resection, embolization therapy, ethanol injection, and microwave coagulation therapy, delay or temporarily control progress, but do not eradicate HCC. Therefore, a new molecular target for anticancer therapy in HCC is needed.
Peroxisome proliferator-activated receptor γ (PPARγ) is a ligand-activated nuclear hormone receptor that mediates the transcriptional regulation of genes. The PPARγ gene is located in human chromosome 3p25; PPARγ includes 4 isoforms: PPARγ1, PPARγ2, PPARγ3, and PPARγ4, which are widely distributed[2]. The PPARγ protein forms a heterodimer with retinoid X receptor α, and the complex subsequently binds to a specific DNA sequence designated as the peroxisome proliferating response element (PPRE), which is located in the promoter region of PPARγ targeted genes and modulates their transcription[3]. Recently, the role of PPARγ in tumors has been extensively studied, and PPARγ agonists have been shown to have direct effects on tumor cells, including breast, colon, lung, stomach, and liver cancer[4,5]. Thiazolidinedione (TZD) groups are synthetic ligands of PPARγ, including troglitazone, rosiglitazone, pioglitazone, and ciglitazone, which have been used widely in patients with insulin-resistant diabetes mellitus. Of note, the highest affinity for PPARγ in the TZD is rosiglitazone[6]. It has been reported that rosiglitazone inhibits the cell proliferation and colony formation via inducing apoptosis in MCF-7 breast cancer cells[7]. However, those effects of rosiglitazone on hepatoma cells have been poorly understood until now.
The phosphatase and tensin homologue deleted on chromosome 10 gene (PTEN) is a major tumor suppressor gene located on human chromosome 10q23.3. The PTEN protein dephosphorylates inositol phospholipid intermediates of the phosphatidylinositol 3-kinase (PI3K) pathway and inhibits the activation of downstream targets including Akt. It has been suggested that the loss of PTEN could stimulate cell proliferation, reduce apoptosis, and induce tumor angiogenesis[8,9]. Previous studies have shown that inactivated PTEN was detected in HCC for its loss, mutation, or low expression[10], and PPARγ activation by rosiglitazone could upregulate the transcription of PTEN in colorectal and breast cancer cells[11]. However, how rosiglitazone affects PTEN in HCC has not been elucidated.
Thus, we hypothesized that the upregulation of PTEN by rosiglitazone could inhibit tumor cell growth, which might be one of the anticancer mechanisms. To verify this hypothesis, the present study investigated the effects of rosiglitazone on the PTEN expression and cell growth in HCC, as well as the underlying mechanisms.
Materials and methods
Reagents Rosiglitazone was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). GW9662, LY294002, and Z-VAD-FMK were obtained from Sigma Chemical Co (St Louis, MO, USA) and were dissolved in 0.1% DMSO. Mouse monoclonal anti-human PTEN, rabbit polyclonal anti-human phosphorylated-Akt (Ser473) and Akt, and horseradish peroxidase-conjugated goat anti-mouse/rabbit IgG secondary antibody were provided by Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture The human HCC cell line Hep3B was kindly donated by Prof Li WEN (Surgical Laboratory Department of Sun-Yat-Sen University, Guangzhou, China). The cells were maintained in the Dulbecco’s modified Eagle’s medium (DMEM, Gibco BRL, Grand Island, NY, USA) containing 10% (v/v) fetal bovine serum (Bio-Whittaker, Walkersville, MD, USA), penicillin (100 U/mL), and streptomycin (100 mg/L). The cells were maintained at 37 °C in an incubator with a humidified atmosphere of 5% CO2.
RNA isolation and RT-PCR The total cellular RNA was extracted using TRIzol reagent (Sigma-Aldrich, USA) according to the manufacturer’s instructions. Reverse transcription with oligo (dT) primer was used to generate cDNA from the total RNA extracts. The PTEN and β-actin genes were amplified using specific sets of primers. The primers for the human PTEN gene were: sense 5'-AGT TTG TGG TCT GCC AGC TA-3', and antisense 5'- TCA GAG TCA GTG GTG TCA GA-3' (470 bp). The primers for the human β-actin gene as an internal control were: sense 5'-GTG GAC ATC CGC AAA GAC-3', antisense 5'-GAA AGG GTG TAA CGC AACT-3' (303 bp). The PCR cycle was as follows: 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 1 min using the TaKaRa Bio Kit (Osaka, Japan) according to the manufacturer’s recommendations with some modifications, and was repeated for 28 cycles. The amplified product was loaded onto 15 g/L agarose gel containing ethidium bromide (0.5 g/L) and quantified by densitometry using the Image Master VDS system and associated software (Pharmacia, Pfizer, NY, USA).
Western blotting analysis The Hep3B cells were rinsed twice with cold PBS buffer, and lysed in an ice-cold lysis buffer including 0.1% SDS, 150 mmol/L NaCl, 50 mmol/L Tris-HCl (pH 7.5), 1% Nonidet P-40, and a protease inhibitor cocktail (Boehringer Mannheim, Lewes, UK) for 30 min at 4 °C. The supernatant was collected by centrifugation at 15 000× g for 20 min. The protein concentration was determined with Coomassie brilliant blue G-250 (Aldrich Chemical, Milwaukee, WI, USA). The cell extracts (50 µg/lane) were separated via SDS-PAGE and electrotransferred to polyvinylidene fluoride membranes (Immobilon, Bedford, MA, USA). After blocking in the blocking buffer (20 mmol/L Tris-HCl, pH 7.6, 150 mmol/L NaCl, 0.1% Tween-20, and 5% non-fat dry milk), the membranes were incubated with primary antibodies overnight at 4 °C and then incubated with horseradish peroxidase-conjugated secondary antibody for 1 h. The blots were developed using an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, Piscataway, NJ, USA) according to the manufacturer’s instructions.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay for cell viability The status of cell growth was determined by MTT (Amresco, Solon, OH, USA) assay. Briefly, exponentially growing cells were diluted to a concentration of 2.5×104 cells/mL with DMEM, planted in 96-well plates (Corning Inc, Corning, NY, USA) with 200 μL/well, and then routinely incubated for 12 h. After being treated with drugs and incubated for 48 h (triplicate wells for each sample), the cells were exposed to 20 µL/well MTT (5 g/L) The medium was then removed after being incubated for 4 h, and DMSO (200 µL/well) was added to dissolve the formazan product. Finally, the plate was read in enzyme-linked immunity implement (Bio-Rad 2550, Hercules, CA, USA) at 570 nm.
Analysis of DNA fragmentation DNA fragmentation was assessed using a modification of a previously described method[12]. Briefly, the Hep3B cells were treated with rosiglitazone at the indicated concentrations and time points, washed with PBS twice, collected, and lysed for 30 min on ice in 400 μL lysis buffer (10 mmol/L Tris-HCl, 10 mmol/L EDTA, and 10 mmol/L NaCl, 1% SDS, pH 8.0). Then an aliquot of the cell lysate was removed for the determination of protein and DNA content. The lysate was treated with 100 µL of proteinase K (500 mg/L) and incubated on ice for 15 min. RNase10 μL (200 mg/L) was added to react for 1 h at 50 °C. DNA was extracted using phenol:chloroform (1:1, v/v) and then loaded onto 1.5% agarose gel containing ethidium bromide (500 mg/L) for electrophoresis. The DNA band was visualized by UV light and photographed.
Flow cytometry analysis 1×106 Hep3B cells treated with rosiglitazone were collected, washed twice with ice-cold PBS, and fixed in 70% ethanol at 4 °C for 12 h. The cells were centrifuged and stained for 30 min with RNase A (50 mg/L) supplemented with propidium iodide (50 mg/L) at 4 °C in the dark. The apoptotic rate was assayed by flow cytometry (EPICS-Elite, Beckman Coulter, Miami, FL USA), and the data were analyzed using CellQuest software (Becton Dickinson, Mountain View, CA, USA). For each sample, 10 000 cells were measured. Hypodiploid cells with a sub-G1 peak (apoptotic peak) were defined as apoptotic cells.
Hoechst 33258 staining assay Apoptotic morphological changes in the Hep3B cells were detected by Hoechst 33258 staining. 1×104 Hep3B cells were washed with ice-cold PBS and fixed with 4% paraformaldehyde in PBS for 10 min at 4 °C. The cells were washed again with PBS and stained with Hoechst 33258 (5 mg/L) for 10 min. The washed cells were then observed under an Advanced Fluorescence Microscope (Nikon 80i, Tokyo, Japan) by an observer blind to the cell treatment.
Detection of caspases activity After the rosiglitazone treatment, the activation of caspases-3, -8, and -9 was detected with 200 µg of the cell lysate using the colorimetric protease assay kit (KeyGEN Biotech, Nanjing, China) according to the manufacturer’s protocol. The activity of caspases-3, -8, and -9 was converted through the measurement of absorbance [optical density (OD) 405 values] using a microplate reader at a wavelength of 405 nm. Non-specific reactions were corrected by subtracting the background absorbance readings from the combination of the cell lysate and buffer.
Gene silencing by small interfering RNA The PTEN small interfering RNA and non-specific control siRNA were purchased from Ribobio (Guangzhou, China). The cells were plated onto 6-well plates, maintained in antibiotic medium for 24 h, and grown to about 50% confluence. PTEN siRNA or control siRNA were transfected with Lipofectamine 2000 reagent (Invitrogen, Madison, WI, USA). Briefly, oligomer-fectamine was diluted at 1:50 in OptiMEMI reduced serum medium (GIBCO, Palo Alto, Calif, USA), mixed gently, and incubated for 5 min at room temperature. Subsequently, a mixture of siRNA was added and incubated for 20 min. The mixture was diluted by adding medium to each well, and the final concentration of siRNA in each well was set as 100 nmol/L. The cells were then incubated for 48 h until processed.
Statistical analysis Data were expressed as mean±SD of at least 3 separate experiments. Differences between the groups were assessed with one-way ANOVA and Student-Newman-Keuls q test using SPSS 11.0 for Windows (SPSS Inc, Chicago, IL, USA). P values less than 0.05 were considered statistically significant.
Results
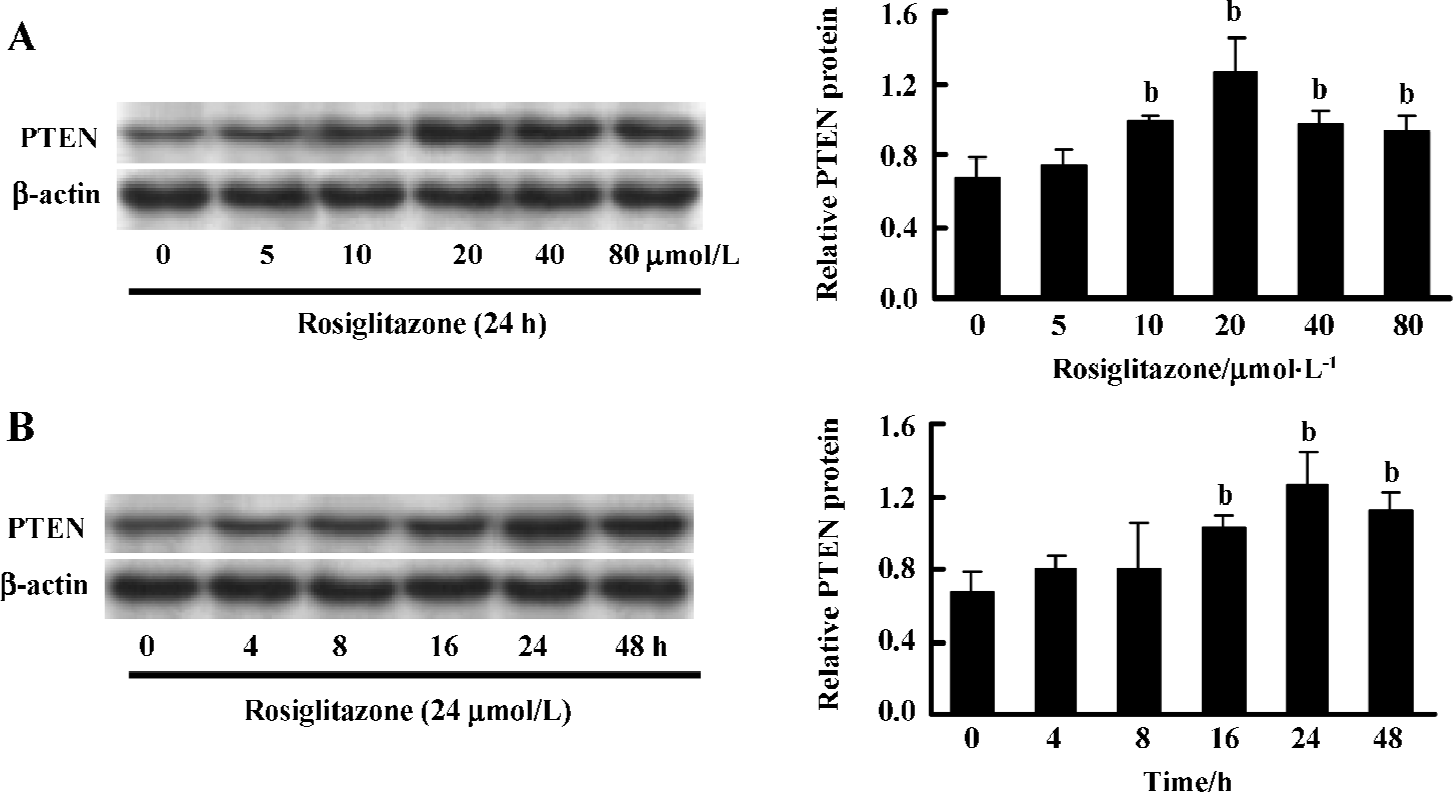
Rosiglitazone stimulated PTEN expression of Hep3B cells in a dose- and time-dependent manner To determine the effect of rosiglitazone on the expression of PTEN protein in HCC cells, the Hep3B cells were treated with various concentrations of rosiglitazone and harvested at different time points. Western blotting showed that rosiglitazone increased the expression level of PTEN in a dose- (Figure 1A) and time-dependent manner (Figure 1B). The most effective dose was 20 µmol/L treated for 24 h. However, the PTEN expression in the Hep3B cells declined slightly after being treated with rosiglitazone for a longer time period (eg 48 h) or at higher concentrations (40 and 80 µmol/L), since the cells were already greatly damaged (data not shown).
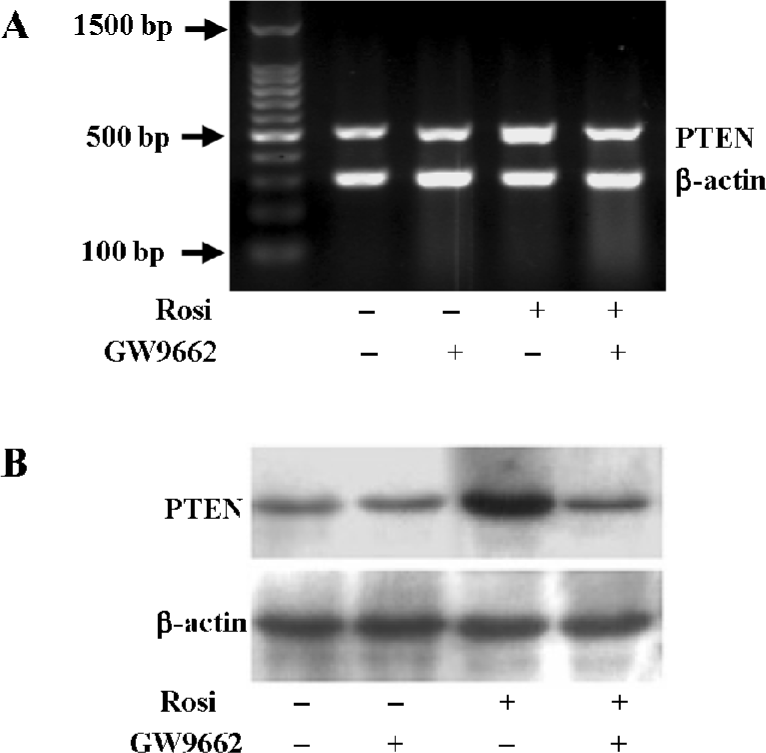
Rosiglitazone increased PTEN expression through the activation of PPARγ To test whether the increase in the PTEN level induced by rosiglitazone was mediated via the activation of PPARγ, the Hep3B cells were pretreated for 1 h with GW9662, a specific and irreversible inhibitor of the PPARγ pathway, and exposed to rosiglitazone for an additional 24 h. Transcription and the expression of PTEN were determined by RT-PCR and Western blotting analyses, respectively. The induced transcription and expression of PTEN were completely blocked in the presence of GW9662, indicating that the increase of the PTEN level was through PPARγ-mediated transcription activation (Figure 2).
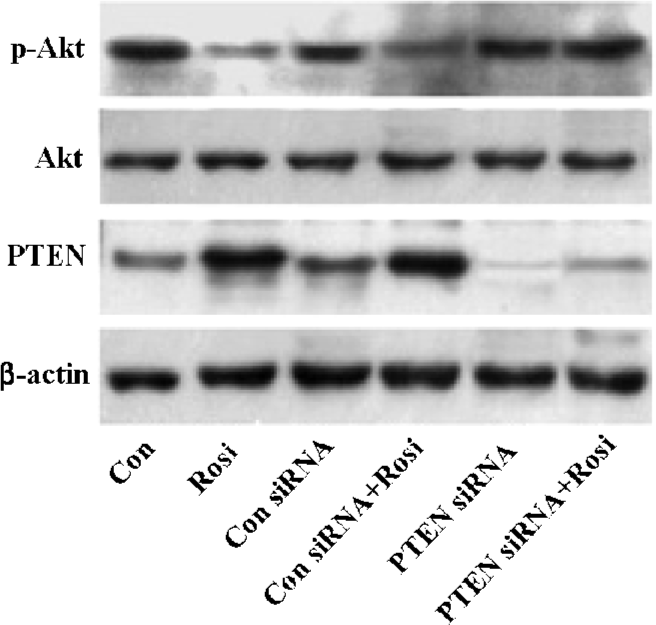
PTEN overexpression induced by rosiglitazone inhibited the PI3K/Akt pathway The oncogene Akt has been confirmed as the key cell survival kinase of the PI3K pathway and is negatively regulated by the tumor suppressor gene PTEN[13,14]. Functional PTEN decreases the Akt phosphorylation level to inhibit the PI3K/Akt pathway[15]. Our results showed that the phosphorylation level of Akt (Ser-473 phosphorylation) was markedly reduced when the expression of PTEN was increased when treated with rosiglita-zone, indicating that rosiglitazone-increased PTEN was a functional protein (Figure 3). To examine whether rosiglitazone had a direct effect on the phosphorylated Akt level, the PTEN gene in the Hep3B cells was silenced with the specific siRNA and treated for 48 h, then the cells were incubated with rosiglitazone for an additional 24 h. Unlike control siRNA, PTEN siRNA almost completely eliminated endogenous PTEN protein production, and the phosphorylated Akt level was not reduced in the presence of rosiglitazone. It is noteworthy that the total Akt protein level did not change (Figure 3). Thus, all data indicated that rosiglitazone affected the phosphorylation level of Akt indirectly and that the reduction in phosphorylated Akt was due to the increase of the PTEN expression. Therefore, rosiglitazone was found to increase active PTEN protein expression, and the PTEN overexpres-sion resulted in the inhibition of the PI3K/Akt pathway activation.
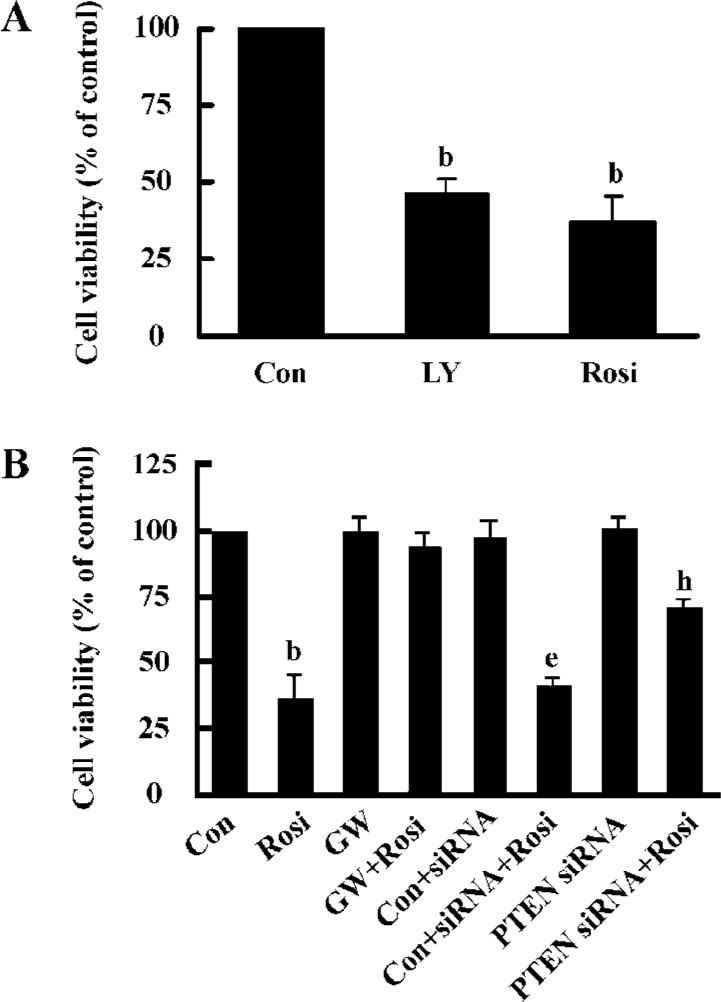
Rosiglitazone inhibited Hep3B cells growth through PPARγ activation Previous reports have demonstrated that Akt plays a critical role in tumorigenesis and cancer progression by stimulating cell proliferation and the inhibition of apoptosis[16,17]. To further evaluate the role of Akt in Hep3B cell growth, LY294002, a specific inhibitor of the PI3K/Akt pathway, was used to block the activation of Akt. When the cells were incubated in the medium containing 30 µmol/L LY294002 or 20 μmol/L rosiglitazone for 48 h, cell viability decreased to 46.6%±4.2% and 37.3%±8.1% of the control, respectively (Figure 4A), which indicated the involvement of activated Akt in Hep3B cell growth. Moreover, GW9662 completely abrogated the inhibitory effect of rosiglitazone on cell growth, suggesting that rosiglitazone induced Hep3B cell death through the PPARγ-dependent pathway (Figure 4B). To evaluate whether PTEN upregulation played a crucial role in the effect of rosiglitazone on cell growth, we silenced the PTEN gene and then the transfected cells were exposed to rosiglitazone for 48 h. The effect of rosiglitazone-decreased cell viability was dramatically reversed, whereas no changes were observed in the control siRNA group (Figure 4B). Those results indicated that the overexpression of PTEN was implicated in the inhibitory effect of rosiglitazone.
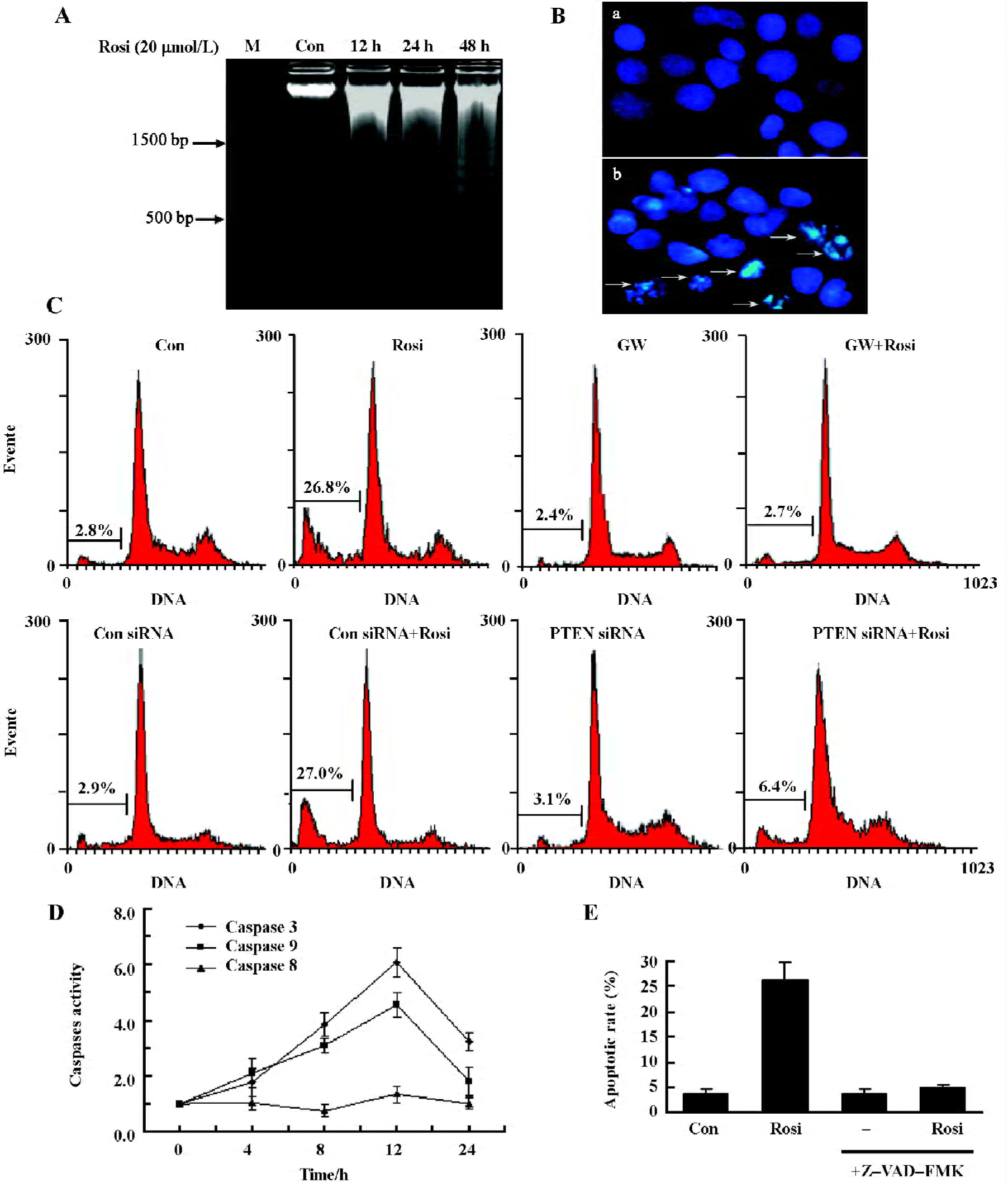
Rosiglitazone-induced apoptosis in cultured Hep3B cells Rosiglitazone can cause apoptosis to Hep3B cells; the DNA ladder was evoked after exposure to 20 μmol/L rosiglitazone for 12, 24, and 48 h (Figure 5A). Hoechst 33258 staining assay drew a similar conclusion: that rosiglitazone (20 µmol/L for 24 h) evoked chromatin condensation in Hep3B cells (Figure 5B). Those findings further supported rosiglitazone-induced apoptosis of Hep3B cells. Moreover, flow cytometry revealed that 20 µmol/L rosiglitazone increased the population of the sub-G1 phase by 26.1%±3.8% for 24 h, and was significantly higher than the 0.1% DMSO-treated control group (Figure 5C). GW9662 completely abrogated the effect of rosiglitazone, suggesting that rosiglitazone induced cell apoptosis via PPARγ activation. Interestingly, the knockdown of PTEN reduced the inductive effect of rosiglitazone on cell apoptosis, indicating that PTEN was required for rosiglitazone-induced apoptosis of Hep3B cells. The activation of PPARγ contributed to 6.4% of the apoptotic rate via other signaling pathways. To further elucidate the underlying molecular mechanism of the apoptosis of Hep3B cells, caspase colorimetric assay was used to assess the effect of rosiglitazone on the activation of caspases-3, -8, and -9 (Figure 5D). Rosiglitazone (20 µmol/L) treatment induced the activation of caspases-3 and -9, and its level reached the peak at 12 h, which was a 6.1- and 4.5-fold increase, respectively. However, the activation of caspase-8 was not found with the same treatment. To evaluate the role of caspases activated in rosiglitazone-induced apopto-sis, we noted that 20 μmol/L Z-VAD-FMK, a pan-caspase inhibitor, completely inhibited caspase activation by rosiglitazone (Figure 5E). Those results suggested that rosiglitazone-induced apoptosis was mediated by the activation of caspases.
Discussion
The main results of our present study first demonstrated that rosiglitazone increased PTEN expression through the activation of PPARγ. PTEN overexpression decreased cell viability and triggered cell apoptosis. Because the loss or downregulation of PTEN and over-proliferation of tumor cells play critical roles in the occurrence and progress of HCC, those effects of rosiglitazone on PTEN and cell growth may be antitumor mechanisms. Rosiglitazone could be a potent chemopreventive or chemotherapeutic agent for treating HCC.
Recently, the PI3K/Akt pathway has become a focus of study. Activated PI3K converts the plasma membrane lipid phosphatidylinositol -4,5-biphosphate (PIP2) to phospha-tidylinositol-3,4,5-triphosphate (PIP3). This phosphorylation then stimulates the catalytic activity of Akt, which affects cell growth, cell cycle entry, and cell apoptosis[18,19]. Previous studies have shown that PTEN is a dual-specificity phosphatase possessing both lipid and protein phosphatase activities. Activated PTEN affects a dephosphorylation of PIP3, generates PIP2, and decreases the phosphorylation level of Akt, which results in cell growth arrest and apopto-sis[20]. PPARγ has been shown to inhibit the transcription of genes related to tumor progression, such as COX-2[21] and NF-κB[22]. We proposed that PPARγ might have a specific relationship with PTEN as a tumor suppressor gene. There-fore, we first confirmed that rosiglitazone upregulated transcription and the expression of PTEN in Hep3B cells. The effect was possibly correlated to PPARγ regulation. Patel et al have found that 2 putative binding sites (PPRE1 and PPRE2) are identified about 10 kb upstream of the minimal promoter region of PTEN[11]. Moreover, it has been reported that rosiglitazone increased PTEN expression in MCF-7 breast cancer[23], AsPC-1 pancreatic cancer[24], non-small-cell lung carcinoma (H1792 and H1838)[25], and HT-29 colon cancer cells[26] through the PPARγ-dependent pathway. Our present results also supported this idea of HCC Hep3B cells.
Functional PTEN inhibits the PI3K/Akt mediated cell survival pathway[20]. We found that Akt activation was required for Hep3B cell growth. This result was consistent with previous reports[27,28]. Rosiglitazone and LY294002 had similar effects on HCC cell death, indicating that their mechanisms in blocking cell growth may be similar. Our data demonstrated that rosiglitazone upregulated the most effective functional PTEN expression to reverse higher levels of Akt phosphorylation in Hep3B cells. The overexpression of PTEN with the treatment of rosiglitazone markedly inhibited Hep3B cell growth, which has been identified as a PPARγ-dependent effect. However, PTEN knockdown did not completely abolish the inhibitory effect of rosiglitazone, suggesting that rosiglitazone could also activate other signaling pathways to inhibit Hep3B cell growth. Thus, further investigation of these effects is needed. Han et al have demonstrated that rosiglitazone inhibits non small cell lung cancer (NSCLC) cell growth not only by the upregulation of PTEN, but also the downregulation of mTOR/p70S6K[25].
Recent reports have shown that the activation of PPARγ strongly induced cell apoptosis of HCC both in vitro and in vivo[29,30]. However, much remains to be learnt about this process. The present results showed that rosiglitazone decreased Hep3B cell viability via cell apoptosis, and the effects were mediated by PPARγ activation. Moreover, we first found the involvement of PTEN upregulation in the apoptotic process of HCC, which might be one of underlying anticancer mechanisms of rosiglitazone. Because PTEN knockdown dramatically reversed the effect of rosiglitazone-induced apoptosis, suggesting that apoptosis induction was not only through the upregulation of PTEN, but also through other signaling pathways, which required further investiga-tion. Of note, the rosiglitazone-induced cleavage of procas-pase to caspases-3 and -9 (Figure 5D) occurred as early as the increase in PTEN expression (Figure 1B) in apoptotic activities. Thus, caspase cleavage might not be absolutely required for PTEN upregulation, at least in the initiation of apoptosis. This also indicates that other pathways via PPARγ activation as early as PTEN upregulation are involved in Hep3B cell apoptosis. This result is consistent with Figure 5C (eighth histogram plot). Because the apoptosis induction via the other pathways had only minor effects (6.4% of apoptotic rate), the PTEN upregulation was considered an important role in the apoptotic process of Hep3B cells. PPARγ activation-mediated apoptosis through c-JNK activation[30] or ERK activation[31] has been described in tumor cell lines, and those effects might be involved in rosiglitazone-induced apoptosis. Interestingly, we further observed that rosiglita-zone induced apoptosis by caspase activation since caspase inhibitor Z-VAD-FMK totally abrogated the effect. In general, 2 main apoptotic pathways are confirmed as the mitochondrial pathway (involving caspase-9) and the death-receptor pathway (involving caspase-8). We found that caspases-9 and -3 were activated by rosiglitazone to produce apoptotic activities. Likewise, previous studies have reported that PPARγ ligands (troglitazone and 15-d-PGJ2) induced apoptosis in hepatoma cell lines through initiating caspases-9 and -3[32,33]. It has been shown that the accumulation of PTEN in the mitochondria mediates rat hippocampal cell apoptosis by inhibiting the PI3K/Akt pathway, the release of cytochrome c, and the activation of caspases-9 and -3[34]. Taken together, those data confirm that rosiglitazone via PPARγ activation induces the Hep3B cell mitochondria apoptosis pathway mainly through the upregulation of PTEN, the inhibition of the PI3K/Akt pathway, and the activation of caspases-9 and -3. Whether rosiglitazone has the same effects in vivo needs to be further observed.
In summary, our results demonstrate that rosiglitazone, a synthetic ligand of PPARγ, increases PTEN expression through the activation of PPARγ, which in turn inhibits the PI3K/Akt pathway and cell growth and induces apoptosis of HCC cells. Additionally, our results suggest that rosiglitazone-induced apoptosis is mediated by caspases-9 and -3 activation. Those observations may explain the underlying mechanisms of the effects of PPARγ agonists on hepatocellular carcinoma, including the effects of rosiglita-zone, and constitute potential novel therapies for the treatment or prevention of liver cancer.
Acknowledgements
The authors appreciate the Surgical Laboratory Department of the First Affiliated Hospital of Sun Yat-Sen University for providing experimental instruments and equipment, and Miao-rong SHE and Xin-hui FU for language editing and technology support.
References
- Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol 2001;2:533-43.
- Koeffler HP. Peroxisome proliferators-activated receptor gamma and cancers. Clin Cancer Res 2003;9:1-9.
- Meirhaeghe A, Tanck MW, Fajas L, Janot C, Helbecque N, Cottel D, et al. Study of a new PPARgamma2 promoter polymorphism and haplotype analysis in a French population. Mol Genet Metab 2005;85:140-8.
- Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res 2003;9:1-9.
- Wang T, Xu J, Yu X, Yang RC, Han ZC. Peroxisome proliferator-activated receptor gamma in malignant disease. Crit Rev Oncol Hematol 2006;58:1-14.
- Young PW, Buckle DR, Cantello BC, Chapman H, Clapham JC, Coyle PJ, et al. Identification of high-affinity binding sites for the insulin sensitizer rosiglitazone (BRL-49653) in rodent and human adipocytes using a radioiodinated ligand for peroxisomal proliferator activated receptor gamma. J Pharmacol Exp Ther 1998;284:751-9.
- Kim KY, Kim SS, Cheon HG. Differential anti-proliferative actions of peroxisome proliferators-activated receptor-gamma agonists in MCF-7 breast cancer cells. Biochem Pharmacol 2006;72:530-40.
- Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI3-kinase signalling via inactivation of PTEN. EMBO J 2003;22:5501-10.
- Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol 2000;20:8969-82.
- Zhang LN, Yu Q, He JY, Zha XL. Study of the PTEN gene expression and FAK phosphorylation in human hepatocarcinoma tissues and cell lines. Mol Cell Biol 2004;262:25-33.
- Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARg agonists are mediated via upregulation of PTEN. Curr Biol 2001;11:764-8.
- Zhou BR, Gumenscheimer M, Freudenberg M, Galanos C. A striking correlation between lethal activity and apoptotic DNA fragmentation of liver in response of D-galactosamine-sensitized mice to a non-lethal amount of lipopolysaccharide. Acta Pharmacol Sin 2003;24:193-8.
- Stoll V, Calleja V, Vassaux G, Downward J, Lemoine NR. Dominant negative inhibitors of signalling through the phosphoinositol 3-kinase pathway for gene therapy of pancreatic cancer. Gut 2005;54:109-16.
- Pedrero JM, Carracedo DG, Pinto CM, Zapatero AH, Rodrigo JP, Nieto CS, et al. Frequent genetic and biochemical alterations of the PI3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer 2005;114:242-8.
- Kreisberg JI, Malik SN, Prihoda TJ, Bedolla RG, Troyer DA, Kreisberg S, et al. Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Res 2004;64:5232-6.
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer 2002;2:489-501.
- Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci USA 2001; 98: 10 983–5.
- Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002;296:1655-7.
- Chan TO, Rittenhouse SE, Tsichlis PN. Akt/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem 1999;68:965-1014.
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidyl-inositol 3,4,5-trisphosphate. J Biol Chem 1998; 273: 13 375–8.
- Yim HW, Jong HS, Kim TY, Choi HH, Kim SG, Song SH, et al. Cyclooxygenase-2 inhibits novel ginseng metabolite-mediated apoptosis. Cancer Res 2005;65:1952-60.
- Okano H, Shiraki K, Inoue H, Yamanaka Y, Kawakita T, Saitou Y, et al. 15-deoxy-delta-12-14-PGJ2 regulates apoptosis induction and nuclear factor-kappaB activation via a peroxisome proliferators-activated receptor-gamma-independent mechanism in hepatocellular carcinoma. Lab Invest 2003;83:1529-39.
- Teresi RE, Shaiu CW, Chen CS, Chatterjee VK, Waite KA, Eng C. Increased PTEN expression due to transcriptional activation of PPARgamma by lovastatin and rosiglitazone. Int J Cancer 2006;118:2390-8.
- Farrow B, Evers BM. Activation of PPARgamma increases PTEN expression in pancreatic cancer cells. Biochem Biophys Res Commun 2003;301:50-3.
- Han S, Roman J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARgamma-dependent and PPARgamma-independent signal pathways. Mol Cancer Ther 2006;5:430-7.
- Chen WC, Lin MS, Bai X. Induction of apoptosis in colorectal cancer cells by peroxisome proliferators-activated receptor gamma activation up-regulating PTEN and inhibiting PI3K activity. Chin Med J (Engl) 2005;118:1477-81.
- Chang HC, Tsai LH, Chuang LY, Hung WC. Role of Akt kinase in sphingosine-induced apoptosis in human hepatoma cells. J Cell Physiol 2001;188:188-93.
- Leng J, Han C, Demetris AJ, Michalopoulos GK, Wu T. Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology 2003;38:756-68.
- Yu J, Qiao L, Zimmermann L, Ebert MP, Zhang H, Lin W, et al. Troglitazone inhibits tumor growth in hepatocellular carcinoma in vitro and in vivo. Hepatology 2006;43:134-43.
- Bae MA, Song BJ. Critical role of c-Jun N-termial protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Mol Pharmacol 2003;63:401-8.
- Li M, Lee TW, Yim AP, Mok TS, Chen GG. Apoptosis induced by troglitazone is both peroxisome proliferator-activated receptor-gamma- and ERK-dependent in human non-small lung cancer cells. J Cell Physiol 2006;209:428-38.
- Toyoda M, Takagi H, Horiguchi N, Kakizaki S, Sato K, Takayama H, et al. A ligand for peroxisome proliferators activated receptor gamma inhibits cell growth and induces apoptosis in human liver cancer cells. Gut 2002;50:563-7.
- Date M, Fukuchi K, Morita S, Takahashi H, Ohura K. 15-Deoxy-delta 12, 14-prostaglandin J2, a ligand for peroxisome proliferators-activated receptor-gamma, induces apoptosis in human hepatoma cells. Liver Int 2003;23:460-6.
- Zhu Y, Hoell P, Ahlemeyer B, Krieglstein J. PTEN: a crucial mediator of mitochondria-dependent apoptosis. Apoptosis 2006;11:197-207.