
Effect of telmisartan on expression of protein kinase C-α in kidneys of diabetic mice1
Introduction
Diabetic nephropathy (DN) is the leading cause of end-stage renal disease (ESRD), accounting for 35% of all new cases in Western countries. Yet the pathogenesis of DN has not been completely understood. Protein kinase C (PKC) is known to be involved in the pathogenesis of DN since it mediates the cellular effects of hyperglycemia, the major trigger of renal impairment of diabetes mellitus (DM)[1]. However, the PKC molecule is not uniformitarian; instead, it consists of 12 subunits with distinct cofactor activation, expression patterns, and cell functions. Their contribution to the pathogenesis of DN is not completely clear. PKC-α, βI, βII, δ, and ζ have been reported to be activated by high-glucose concentration in various cell culture models and in the kidneys of diabetic rats[2,3]. Kang et al reported that PKC-α was markedly increased in renal glomeruli and interstitial capillaries as well as in the endothelial cells of large arteries in streptozotocin (STZ)-induced diabetic rats while other isoforms were less distinctly affected[4]. Recently, Thallas-Bonke et al showed that PKC-α was especially stimulated by advanced glycation end products in the diabetic kidney[5]. These data indicate the prominent role PKC-α in DN. More recently, PKC-α knockout mice were established and their renal phenotypes were partly investigated by us and other groups[6,7]. To further elucidate the role of PKC-α in the pathophysiology of DN with this knockout-mouse model, we investigated the expression and distribution of PKC-α, which proved to be different between normal mice and rats, in the kidneys of diabetic mice.
Both transforming growth factor-β1 (TGF-β1) and vascular endothelial growth factor (VEGF) have been implicated in the pathophysiology of DN. TGF-β1 is involved in the development of mesangial matrix deposition, glomerular basement membrane (GBM) thickening, and promoting podocyte apoptosis or detachment in DN[8–10], whereas VEGF takes part in the regulation of glomerular permeability, glomerular endothelial cell growth, and urinary albumin excretion[11–13]. The enhanced expressions of TGF-β1 and VEGF stimulated by high glucose have proven to be regulated via a PKC-dependent pathway[14–16]. A recent study showed that PKC-α lead to an increased expression of TGF-β1[17], but other studies have reported that PKC-α is only involved in the regulation of VEGF and its receptor rather than TGF-β1 under high glucose conditions[18]. Therefore, the relations of PKC-α to TGF β1 and VEGF need further clarification.
It has been demonstrated that angiotensin (Ang) II is involved in almost every pathophysiological process implicated in the development of DN, including hemodynamic changes, hypertrophy, extracellular matrix accumulation, growth factor/cytokine induction, reactive oxygen species (ROS) formation, podocyte damage, proteinuria, and interstitial inflammation[19]. Experimental and clinical studies have established that blocking the rennin Ang system (RAS) reduces the glomerular filtration of proteins and delays the deterioration of renal function in DN[20,21]. However, the molecule events that underlie the impact of RAS remain unclear. PKC has been suggested to mediate the effects of RAS. For example, Ang II was suggested to stimulate the transport of Na+ in proximal tubules mediated by PKC activation. Considering that PKC-α gene knockout DN mice exhibited low urinary protein excretion[6], we assume that blocking RAS might influence the expression and/or activation of PKC-α in the kidneys of diabetic mice, which could contribute to their renoprotective effects.
Materials and methods
STZ, Cy3 conjugated goat anti-rabbit IgG, rabbit anti-mouse TGF-β1, and VEGF antibodies were purchased from Sigma (St Louis, MO, USA). Proteinase K, donkey anti-rabbit IgG conjugated with fluorescein isothiocyanate (FITC) and donkey anti-goat IgG conjugated with Texas Red were purchased from San-Ying Co (Wuhan, China). FITC-conjugated phalloidin was purchased from Alexis Co (San Diego, CA, USA). Rabbit anti goat PKC-α and, goat anti mouse Tamm-Horsfall, and aquaporin (AQP)-2 antibodies were purchased from Santa Cruz Co (San Diego CA, USA). A protein ELISA kit was purchased from Roche Co (Karlsruhe, Germany). Ang II receptor blocker (ARB) telmisartan was kindly provided by Boehringer Ingelheim Pharmaceuticals Inc Co (Shanghai, China). Major apparatus included the cryosection machine (Leica CM3050S, Nussloch Germany) and confocal laser-scanning microscopy (Olympus FV500, Tokyo, Japan).
Animals C57BL/6 male mice (25–35 g) were purchased from Laboratary of Transplantation Center of Tongji Hospital(Wuhan, China). All of the experiments were in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals. The diabetic mice were induced by an intraperitoneal injection of STZ (100 mg/kg body weight dissolved in cold citrate buffer, pH 4.0) every other day. One week later, the glucose concentration was determined in tail blood samples, and only the mice with blood glucose above 16.7 mmol/L were considered diabetic. The non-diabetic mice initially injected with STZ vehicle served as controls (group N, n=6). The diabetic mice then received telmisartan (9 mg·kg-1·d-1 po, group T, n=7) or vehicle (group DN, n=7) by gastric tube. During the whole experiment, the mice had free access to standard mouse chow and tap water. Two mice, each from the DN and T groups, died on d 21 and 26, respectively, after the last administration of STZ. On d 41, 24 h urine samples were collected in metabolic cages. The protein concentration in the urine was determined by Coomassie blue G binding assay for protein with bovine serum albumin as standard. Forty-two days after the application of STZ or vehicle, the following experiments were performed on the surviving mice.
Preparation of kidney samples The experimental mice were anesthetized by intraperitoneal injection of pentobarbital sodium (70 mg/kg). After opening the abdominal cavity, the left renal artery was clamped and the left kidney was excised. The cortex and the outer and inner medulla were dissected and immediately immersed into liquid nitrogen for Western blot analysis; then the heart was exposed. The tip of the perfusion system was inserted into the left ventricle, and the arterial system was perfused for 1 min with 3–5 mL PBS (120 mmol/L NaCl, 16 mmol/L Na2HPO4, and 2.9 mmol/L KH2PO4) to clear the blood of the kidney, and subsequently for 10 min with 10–15 mL fixation solution (4% paraformaldehyde and 3% sucrose in phosphate buffer). Both solutions were at room temperature. Then the right kidney was removed and weighed for immunohistochemical and morphological examinations.
Preparation of cryosections The kidneys were cut into slices (3 mm thickness) displaying the cortex and outer and inner medulla. Parts of kidney tissues were fixed with 10% neutral formalin, imbedded with paraffin, cut into slices (2 μm thickness), and mounted onto gelatin-coated, glass slides. The remaining tissues were incubated for 15 min in the fixation solution at 4 ℃. After rinsing in PBS for 15 min, the kidney slices were dehydrated in 30% sucrose in PBS for 6 h at 4 ℃. Thereafter, the kidney slices were frozen in isopentane, precooled by liquid nitrogen and stored at -80 ℃ until further use. Cryosections of ~8 µm were made at -20 ℃ and transferred onto gelatin-coated, glass slides.
Hematoxylin and eosin (HE) staining and glomeruli morphometry The sections were deparaffinized with xylene, rehydrated in graded ethanol, and then stained with periodic acid-schiff (PAS). Three cryosections of each kidney were randomly chosen and stained with HE. Thirty glomeruli were randomly selected from the sections of each subject at an original magnification of ×400. The following parameters were determined from light microscopic images using the HMIAS-2000 system (Qian-Ping Co, Wuhan, China): the total cell number of glomeruli, mean glomeruli area, and perimeter.
Immunohistochemistry Immunofluorescence staining and confocal laser scanning microscopy were performed as previously described[21]. All steps were performed at room temperature. Briefly, after pre-incubation for 20 min in PBS containing 4% normal goat serum or normal donkey serum plus 1% bovine serum albumin and 0.25% Triton X-100, the cryosections were incubated for 1.5 h with rabbit anti goat PKC-α, TGF-β1, and VEGF antibodies (1 µg/mL). For the negative control, the primary antibodies were pre-absorbed with their corresponding peptides by incubating them with a 10-fold excess of peptide antigen in PBS for 2 h at room temperature before the incubation of the sections with the antibody-peptide solution. After the sections were washed 3 times for 5 min in PBS, they were incubated for 1.5 h with donkey anti-rabbit IgG conjugated with Cy3 (final concentration 5 µg/mL; Sigma, USA). The sections were then washed twice for 10 min in PBS and mounted in FluorSave (Calbiochem, San Diego, CA, USA) as a fading retardant. The glomerular fluorescence intensity was determined by Image-Pro Plus version 5.0 System (Media Cybernetics Inc, Georgia, USA).
Double-staining experiments were performed with PKC-α antibodies and a primary antibody against the respective tissue epitope. For the labeling of filamentous actin in the foot process of podocytes and proximal tubular brush border, FITC-conjugated phalloidin (200 U/mL) was used. The thick ascending limb of Henle’s loop was labeled by a polyclonal antibody (goat antiserum) against the Tamm-Horsfall protein (2 µg/mL, ICN, Eschwege, Germany). To study the expression of PKC in the cortical and medullary-collecting duct, a polyclonal antibody against AQP-2 (2.0 µg/mL) was applied. The following secondary antibodies (5µg/mL) were used: donkey anti-rabbit IgG conjugated with FITC or goat anti-rabbit IgG conjugated with Cy3 for PKC antibodies, and donkey anti-goat IgG conjugated with Texas Red for the anti-AQP2 antibody and for the antibody against the Tamm-Horsfall protein. The sections were studied by confocal laser-scanning microscopy.
Western blot analysis Western blot analysis was as described previously[7]. The total cellular proteins of the cortex and the outer and inner medulla were obtained separately by pulverizing the tissue and dissolving the powder in lysis buffer. Homogenization was followed by centrifugation (1.000×g, 10 min at 4 °C). The protein content was determined by the Bradford method, using bovine serum albumin as standard. Samples were diluted 1:3 with Roti-Load sample buffer and boiled for 10 min at 65 ℃. The following steps were performed at room temperature: samples of 10 µg protein were subjected to SDS gel electrophoresis using 10% acrylamide gels in a Mini-PROTEAN II Electrophoresis Cell (Bio-Rad Co, München, Germany). For the determination of molecular weight, a prestained protein ladder was used. After gel electrophoresis (60 mA/gel, 70 min), the proteins were transferred to the polyvinylidene-fluoride (PVDF) membrane of 0.45 µm pore size. The membranes were blocked for 90 min with blocking buffer and rinsed twice with triethanolamine-buffered saline (TBS) containing 0.1% Tween 20 (TBST). Thereafter, the PKC-α antibodies (0.5 µg/mL) were incubated overnight in TBST. The secondary horseradish peroxidase-conjugated antibody was incubated at a concentration of 0.1 µg/mL for 2 h, RT in TBST. The blots were rinsed twice with TBST and washed 3 times for 15 min with TBST, then 2 times in assay buffer at RT. Immunoreactive proteins were detected using enhanced chemiluminescene (ECL)-Western blotting detection system according to the manufacturer’s instructions. The membranes were exposed to Hyperfilm-ECL autoradiography films (Amersham biosciences, Buckinghamshire, UK) for 2 min and quantified with the image-analysis system MGIAS-1000 (Bio-Rad Co, München, Germany).
Statistical analysis Data are presented as mean±SEM. Statistical analysis was carried out by means of one-way ANOVA using SPSS 10.0 (SPSS Inc Chicago, USA). Pearson’s correlation analysis was used to demonstrate correlations between the data. For all analyses, values of P<0.05 were considered statistically significant.
Results
General characterization of STZ-induced diabetic mice More than 16.7 mmol/L blood glucose persisted in STZ-induced diabetic mice during the 6 week study period, which could not be attenuated by telmisartan treatment. Body weight, right kidney weight, right kidney-to-body weight ratio, and 24 h urinary protein excretion of STZ-induced diabetic mice were significantly higher than the controls (P<0.05); these changes could be attenuated by telmisartan treatment (P<0.05 vs normal and DN mice) (Table 1).

Full table
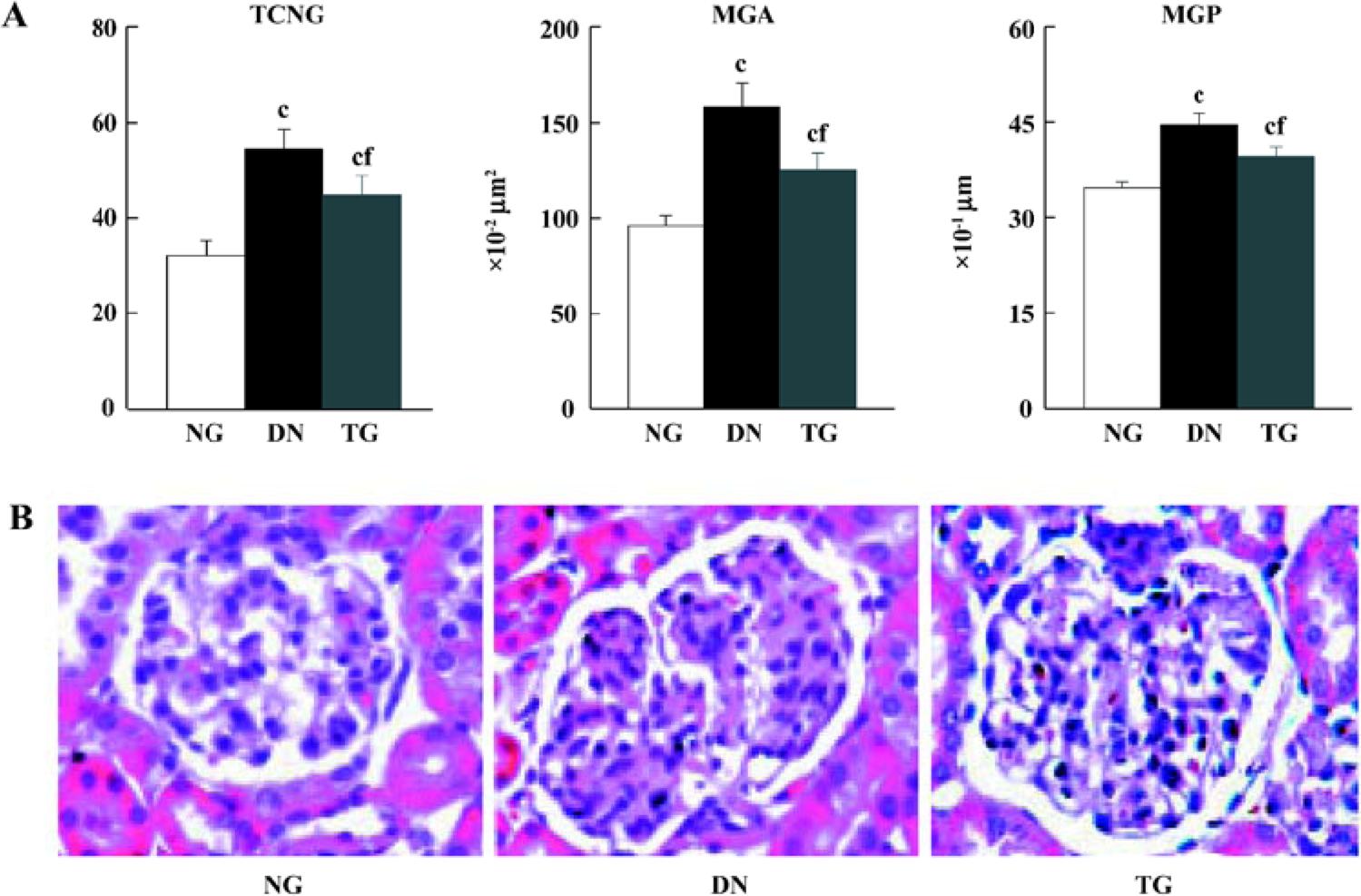
HE staining and morphometric analysis HE staining revealed a significant increase in mean glomeruli area, perimeter, and total cell number in the kidneys of STZ-induced diabetic mice compared to the controls (P<0.05). These pathological changes could partly be inhibited by telmisartan treatment (P<0.05 vs normal and DN mice) (Figure 1).
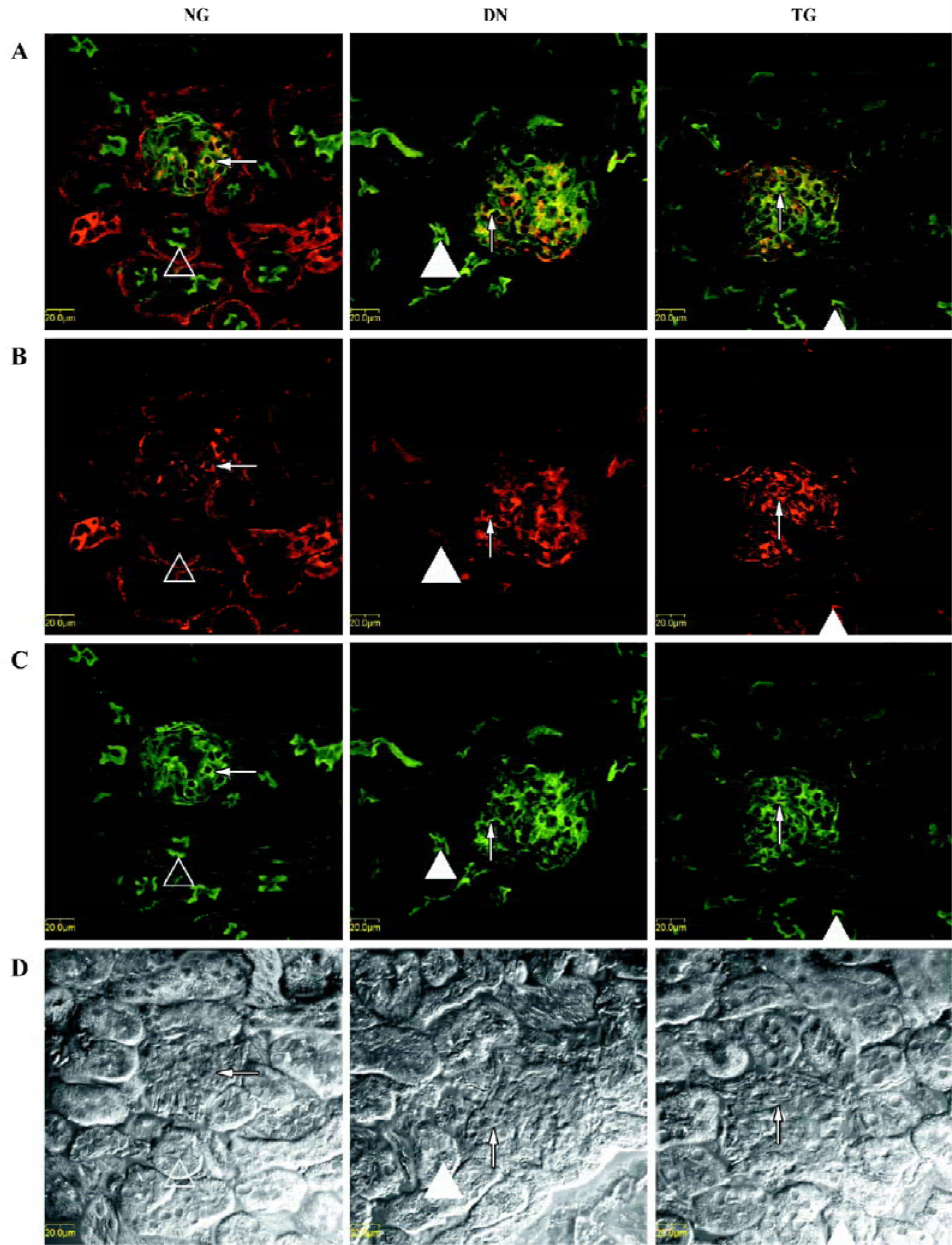
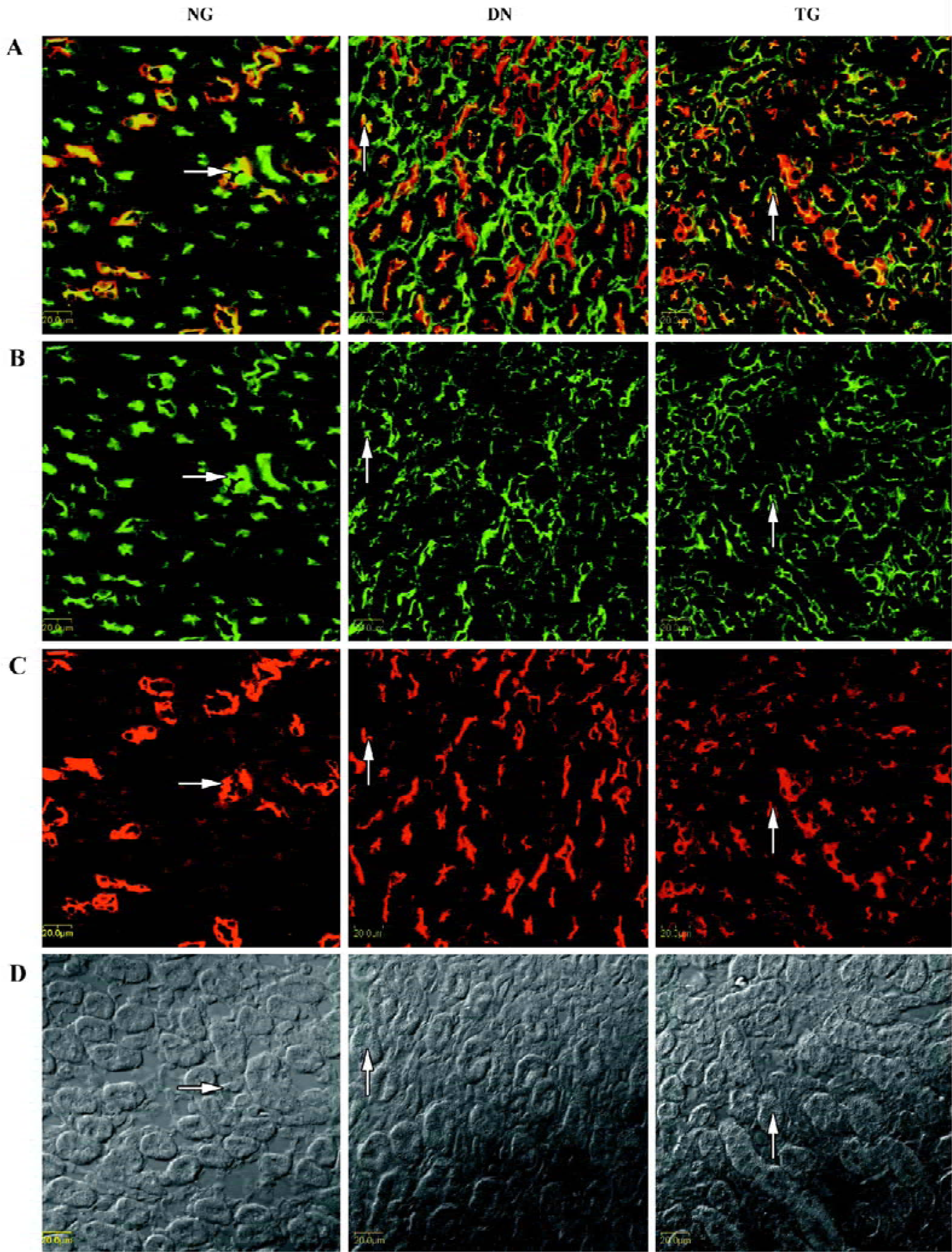
Immunohistochemistry The distributions of PKC-α in the kidney of normal, diabetic, and telmisartan-treated diabetic mice are depicted in Table 2 and illustrated in part in Figures 2 and 3. In normal mice, immunostaining for PKC-α was detected in the podocyte of glomeruli, the epithelial cells of proximal tubules, and medullary-collecting duct (MCD). There was no significant signal detectable in the medullary and cortical thick ascending limb (data not shown). The expression in other glomeruli structures, such as the mesangial cell-like structure, could not be excluded. The diabetic mice exhibited similar renal distribution of PKC-α except for the translocation from the basement membrane to the apical membrane of epithelial cells in proximal tubules and from the apical membrane to the basement membrane of epithelial cells in the inner medullary-collecting duct (IMCD). Telmisartan treatment could attenuate these changes.
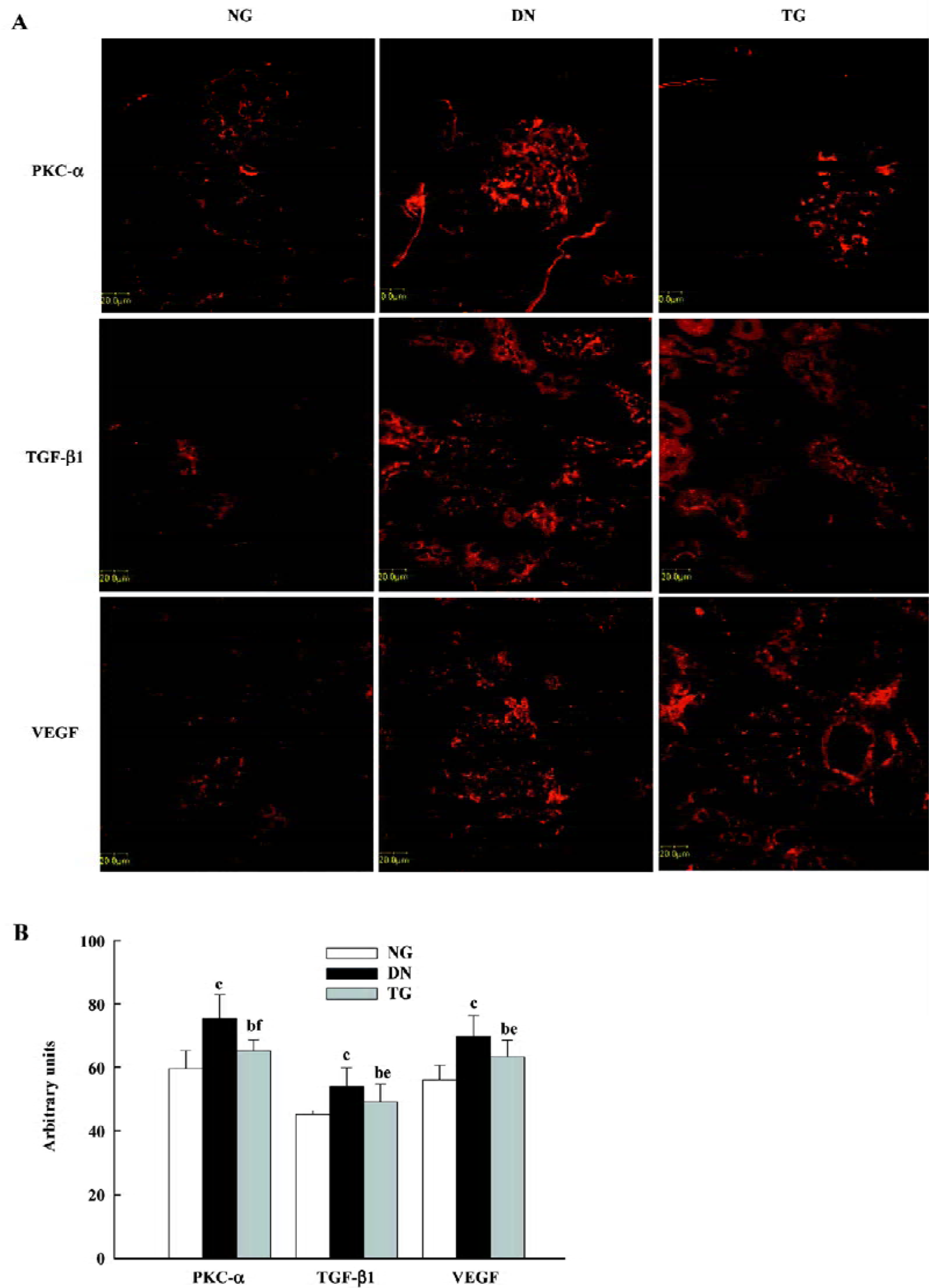
In addition, the enhanced glomerular expressions of PKC-α, TGF-β1, and VEGF were detected by semiquantitative immunostaining analysis, and the application of telmisartan appeared to ameliorate these changes (P<0.05 vs normal and DN groups) (Figure 4). Pearson’s correlation analysis showed a positive correlation of PKC-α to VEGF (r=0.600, P=0.039), but no correlation to TGF-β1 (r=0.432, P=0.054).
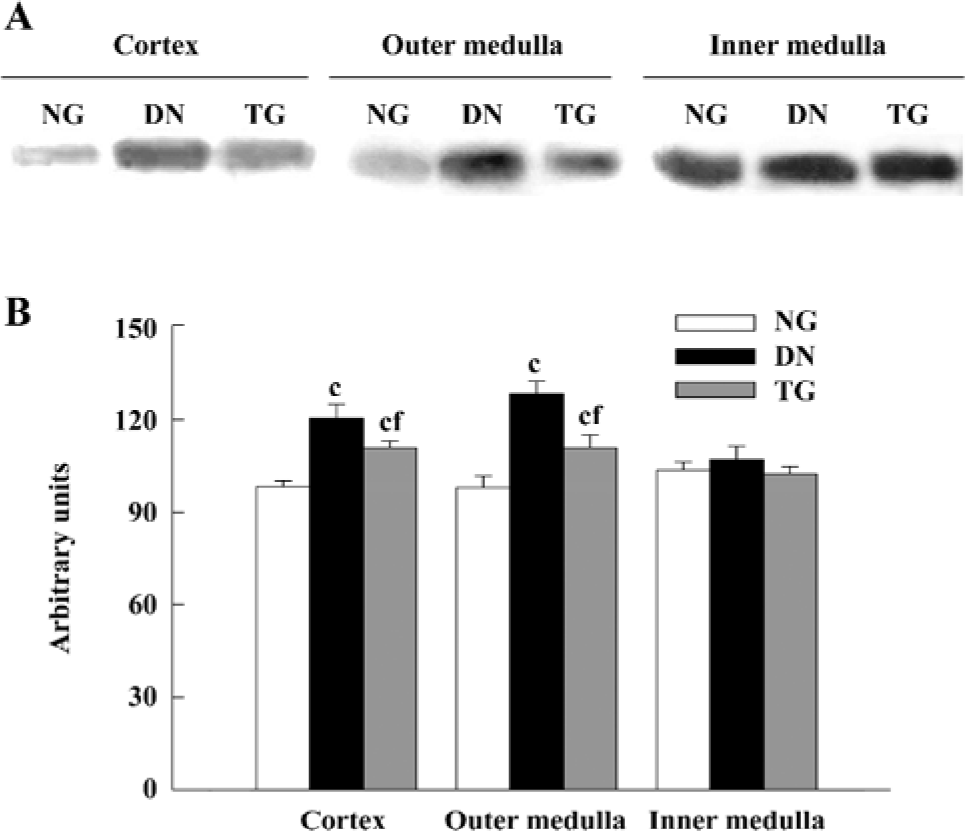
Western blot analysis The expressions of PKC-α in the cortex and outer and inner medulla of the kidneys were determined separately. The PKC-α antibody recognizes 80 kDa bands. Western blot analysis detected an enhanced expression of PKC-α in the renal cortex and outer medulla of diabetic mice (P<0.05 vs normal group), while in the telmisartan-treated diabetic mice, such enhanced expression was decreased significantly (P<0.05 vs normal and DN groups) (Figure 5).
Discussion
STZ-induced diabetic mice persisted with high blood glucose until the whole experiment was finished. Similar to clinical DN, these diabetic mice had higher body weight, right kidney weight, right kidney-to-body weight ratio, and 24 h urinary protein excretion than the normal control. HE staining further verified that the pathology changes of these mice models were in accordance with those of the clinical DN stage. With confocal laser scanning microscopy, we found the expression of PKC-α in glomeruli, epithelial cells of proximal tubules, and MCD of DN mice, but not in the medullary and cortical thick ascending limb. Such a distribution of PKC-α is similar to normal mice, but different from normal rats in the thick ascending limb as reported by Redling et al[22,23].
However, in epithelial cells of proximal tubules of DN mice, PKC-α was mostly translocated from the basement membrane to the apical membrane, which is considered to be an important sign of PKC-α activation[24], whereas PKC-α was largely translocated from the apical membrane to the basement membrane in epithelial cells of IMCD, which denotes decreased activation of PKC-α. These important findings indicate the different states of activation of PKC-α may occur in different renal tissues under the same DN condition. In fact, we also observed in the study that the expression of PKC-α increased in the renal cortex and outer medulla, but not in the inner medulla of DN mice. Undoubted-ly, such different states of activation, as well as expression, should imply different roles and mechanisms of regulation of PKC-α in different structures of the kidneys in DN mice.
The observation of enhanced activation of PKC-α in renal proximal tubules of DN mice in vivo is in accordance with the in vitro report by Karim et al[25]. Some investigators have demonstrated that PKC-α activation is critical for the inhibition of Na+-K+-ATPase in epithelial cells of proximal tubules in vitro[26,27], which is believed to contribute to the pathogenesis of DM and its complications[28]. In addition, PKC-α activation was also suggested to be involved in albumin uptake in proximal tubules in vitro[29]. Therefore, enhanced activation of TGF-β1 in renal proximal tubules may have been associated with function changes in renal proximal tubules of DN mice. On the other hand, we found, for the first time, decreased activation of PKC-α in IMCD in DN mice. So far, few studies have reported the activity of PKC-α in IMCD, thus little is known about its implication. Nevertheless, our recent study demonstrates that PKC-α-deficient mice exhibit impaired urinary concentrating function due to the defect in IMCD[7]. So such decreased activation of PKC-α in IMCD is supposed to be responsible for defective urine concentrating function in DN mice. However, the mechanisms as to how PKC-α contributes to urinary concentration under DN conditions, especially the signaling pathway of its activation or inactivation and its targets, still needs to be investigated.
Most previous reports focused on the activation of PKC. In contrast, we observed increased expression of PKC-α in renal glomeruli, which supports a previous report on the expression of PKC-α, which mainly affected the kidneys of diabetic rats[4]. Kang et al suggested that the enhanced expression of PKC might underlie the sustained PKC activation observed in diabetes and hyperglycemia[4]. However, little evidence is available to support such a hypothesis so far. Recently, the links between PKC-α and TGF-β1 or VEGF have been proposed. In accordance, we found that the expressions of TGF-β1 and VEGF were all increased as well as PKC-α. Moreover, the increased expression of PKC-α only correlated to VEGF rather than TGF-β1, which was in accordance with a recent study where PKC-α-deficient mice were observed[6]. Therefore, despite the lack of direct evidence, we postulate that the enhanced expression ofPKC-α in renal glomeruli may be involved in the pathogenesis of DN, at least in association with the increased expression of VEGF.
It is well known that most of the intrarenal effects of Ang II are mediated by Ang II type 1 receptor (AT-1R), which can be inhibited by ARB producing renoprotective effects, while the beneficial effects of ARB on the diabetic renal structural and functional changes in turn provide the evidence for the role of molecules involved in DN. Recently, the cardiac RAS was reported to promote PKC translocation in the diabetic heart via AT-1R, indicating the role of AT-1R in connecting RAS to PKC, but which PKC isozyme is involved was not shown[30]. In the present study, we found proteinuria and right kidney-to-body weight ratio were significantly ameliorated after the treatment of DN mice with ARB telmisartan. Meanwhile, the enhanced expression of PKC-α as well as VEGF in renal glomeruli was attenuated. The different translocations of PKC-α in renal proximal tubules and IMCD were also significantly reduced after treatment with telmisartan. These data not only indicate that the nephroprotective effects of ARB telmisartan may be mediated more or less by PKC-α, but also suggest that PKC-α may play a role in the RAS-PKC signaling cascade in DN.
Taken together, we mainly found the enhanced expression of PKC-α in renal glomeruli, increased activation of PKC-α in renal proximal tubules, and for the first time, decreased activation of PKC-α in IMCD of DN mice. Combined these with the effects of ARB telmisartan, our findings suggest that PKC-α may play a role in the pathogenesis of DN, and that nephroprotective effects of ARB telmisartan may be partly associated with its influence on PKC-α.
References
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001;414:813-20.
- Haller H. Postprandial glucose and vascular disease. Diabet Med 1997;14:S50-6.
- Meier M, King GL. Protein kinase C activation and its pharmacological inhibition in vascular disease. Vasc Med 2000;5:173-85.
- Kang N, Alexander G, Park JK, Maasch C, Buchwalow I, Luft FC, et al. Differential expression of protein kinase C isoforms in streptozotocin-induced diabetic rats. Kidney Int 1999;56:1737-50.
- Thallas-Bonke V, Lindschau C, Rizkalla B, Bach LA, Boner G, Meier M, et al. Attenuation of extracellular matrix accumulation in diabetic nephropathy by the advanced glycation end product cross-link breaker ALT-711 via a protein kinase C-alpha-dependent pathway. Diabetes 2004;53:2921-30.
- Menne J, Park JK, Boehne M, Elger M, Lindschau C, Kirsch T, et al. Diminished loss of proteoglycans and lack of albuminuria in protein kinase C-α-deficient diabetic mice. Diabetes 2004;53:2101-9.
- Yao L, Huang DY, Pfaff IL, Nie X, Leitges M, Vallon V. Evidence for a role of protein kinase C alpha in urine concentration. Am J Physiol Renal Physiol 2004;287:F299-304.
- Chen S, Hong SW, Iglesias-de la Cruz MC, Isono M, Casaretto A, Ziyadeh FN. The key role of the transforming growth factor-β system in the pathogenesis of diabetic nephropathy. Ren Fail 2001;23:471-81.
- Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 2005;54:1626-34.
- Chen S, Jim B, Ziyadeh FN. Diabetic nephropathy and transforming growth factor-beta: transforming our view of glomerulosclerosis and fibrosis build-up. Semin Nephrol 2003;23:532-43.
- De Vriese AS, Tilton RG, Elger M, Stephan CC, Kriz W, Lameire NH. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol 2001;12:993-1000.
- Flyvbjerg A, Dagnaes-Hansen F, De Vriese AS, Schrijvers BF, Tilton RG, Rasch R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes 2002;51:3090-4.
- Schrijvers BF, Flyvbjerg A, De Vriese AS. The role of vascular endothelial growth factor (VEGF) in renal pathophysiology. Kidney Int 2004;65:2003-17.
- Cha DR, Kim NH, Yoon JW, Jo SK, Cho WY, Kim HK, et al. Role of vascular endothelial growth factor in diabetic nephropathy. Kidney Int Suppl 2000;77:S104-12.
- Williams B, Gallacher B, Patel H, Orme C. Glucose-induced protein kinase C activation regulates vascular permeability factor mRNA expression and peptide production by human vascular smooth muscle cells in vitro. Diabetes 1997;46:1497-503.
- Hoshi S, Nomoto K, Kuromitsu J, Tomari S, Nagata M. High glucose induced VEGF expression via PKC and ERK in glomerular podocytes. Biochem Biophys Res Commun 2002;290:177-84.
- Lindschau C, Quass P, Menne J, Guler F, Fiebeler A, Leitges M, et al. Glucose-induced TGF-β1 and TGF-β receptor-1 expression in vascular smooth muscle cells is mediated by protein kinase C-α. Hypertension 2003;42:335-41.
- Niimi Y, Mochida S, Matsui A, Inao M, Fujiwara K. PKC- and MAPK-independent upregulation of VEGF receptor expressions in human umbilical venous endothelial cells following VEGF stimulation. Hepatol Res 2001;21:261-7.
- Wolf G. New insights into the pathophysiology of diabetic nephropathy: from haemodynamics to molecular pathology. Eur J Clin Invest 2004;34:785-96.
- Makino H, Haneda M, Babazono T, Moriya T, Ito S, Iwamoto Y, et al. The telmisartan renoprotective study from incipient nephropathy to overt nephropathy-rationale, study design, treatment plan and baseline characteristics of the incipient to overt: angiotensin II receptor blocker, telmisartan, Investigation on Type 2 Diabetic Nephropathy (INNOVATION) Study. J Int Med Res 2005;33:677-86.
- Wienen W, Richard S, Champeroux P, Audeval-Gerard C. Comparative antihypertensive and renoprotective effects of telmisartan and lisinopril after long-term treatment in hypertensive diabetic rats. J Renin Angiotensin Aldosterone Syst 2001;2:31-6.
- Redling S, Pfaff IL, Leitges M, Vallon V. Immunolocalization of protein kinase C isoenzymes α, βI, βII, δ, and ζ in mouse kidney. Am J Physiol Renal Physiol 2004;287:289-98.
- Pfaff IL, Wagner HJ, Vallon V. Immunolocalization of protein kinase C isoenzymes α, βI and βII in rat kidney. J Am Soc Nephrol 1999;10:1861-73.
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992;258:607-14.
- Karim Z, Defontaine N, Paillard M, Poggioli J. Protein kinase C isoforms in rat kidney proximal tubule: acute effect of angiotensin II. Am J Physiol Cell Physiol 1995;269:134-40.
- Khundmiri SJ, Dean WL, McLeish KR, Lederer ED. Parathyroid hormone-mediated regulation of Na+-K+-ATPase requires ERK-dependent translocation of protein kinase Calpha. J Biol Chem 2005;280:8705-13.
- Liang M, Knox FG. Nitric oxide activates PKC-alpha and inhibits Na+-K+-ATPase in opossum kidney cells. Am J Physiol 1999;277:F859-65.
- Bagrov YY, Manusova NB, Egorova IA, Fedorova OV, Bagrov AY. Endogenous digitalis-like ligands and Na/K-ATPase inhibition in experimental diabetes mellitus. Front Biosci 2005;10:2257-62.
- Hryciw DH, Pollock CA, Poronnik P. PKC-alpha-mediated remodeling of the actin cytoskeleton is involved in constitutive albumin uptake by proximal tubule cells. Am J Physiol Renal Physiol 2005;288:F1227-35.
- Malhotra A, Kang BP, Cheung S, Opawumi D, Meggs LG. Angiotensin II promotes glucose-induced activation of cardiac protein kinase C isozymes and phosphorylation of troponin I. Diabetes 2001;50:1918-26.