
High-throughput real-time assay based on molecular beacons for HIV-1 integrase 3'-processing reaction1
Introduction
Human immunodeficiency virus (HIV) integrase (IN) is a validated and an important target for the development of novel drugs for several reasons. First, it is an essential enzyme in the retroviral life cycle[1,2]. The integration of the proviral DNA into transcriptional active sites of the host DNA represents a point of no return[3]. Second, it has no cellular homologue[4,5]. Drugs selectively targeting IN are expected to show low toxicity in the host. Third, X-ray crystal and Nuclear magnetic resonance (NMR) structures of the catalytic domain of HIV IN are available for rational structure-based drug design[6–8]. In addition, the combination of IN inhibitors with those directed against protease and reverse transcriptase was shown to be synergistic in several tested models[9]. IN can catalyze 2 specific enzymatic reactions known as 3'-processing and DNA strand transfer[1,10]. In the first step, IN removes 2 nucleotides from each 3' end of the linear viral DNA made by reverse transcription. This step is sequence specific and dependent on the recognition of a highly conserved 5'-CAGT-3' site in the U3 and U5 regions of the viral long terminal repeats (LTR). In the subsequent strand transfer reaction, the 3'-hydroxyl group of each cDNA strand attacks a strand of the cellular DNA and splices these 3' ends into the host chromosome.
Both the 3'-processing and strand transfer reactions can be carried out in vitro with purified recombinant IN, a divalent metal cation such as Mg2+ or Mn2+, a donor oligonucleotide that mimics 1 of the viral DNA ends, and a target oligonucleotide[11–20]. Traditionally, IN activity is measured by a radiolabeled substrate assay in which reaction products are separated on polyacrylamide denaturing gels and subjected to autoradiography[1,11,12]. The assay is highly sensitive and commonly used for inhibitor screening[11–13]. However, the disadvantages of the method are that: (i) it requires radiolabeled substrates, special equipment, and appropriate handling of hazardous radioactive waste; and (ii) it is inconvenient to process a large amount of reactions due to the low-throughput format. Compared with the radiolabeled substrate assay, biotin (or digoxin)-labeled microtiter plate assays are safe and high-throughput, and the products of IN reactions are easy to measure with a spectrophotometer[14–16]. However, the major drawbacks of these microtiter plate assays are the low signal/noise ratio and intensive labor because of the necessity for plate coating and repeated washing. Fluorescence technology, with the advantages of high specificity and sensitivity, has become an important tool for studying nucleic acids and protein/DNA interactions. Recently, some fluorescent-based assays were developed as high-throughput screening protocols for IN reactions[17–20].
A new fluorescence technique, known as molecular beacons, was reported by Tyagi and Kramer in 1996 for the construction of probes that are useful for real-time detection of nucleic acids in homogenous solutions[21]. Those probes are hairpin-shaped oligonucleotides, each with a fluorophore covalently linked to 1 end and a quencher moiety covalently linked to the other end. The fluorophore is quenched by energy transfer when the stem keeps the ends in close proximity. When the probe undergoes a spontaneous conformational change forcing the stem apart, fluorescence occurs. Recently, molecular beacons have been widely used in the investigation of DNA-protein interactions including DNA cleavage and ligation reactions because of their high sensitivity, simplicity, and real-time monitoring[22–25]. In this study, we describe a new high-throughput assay that translates the IN 3'-processing reaction into a fluorescent signal in real-time using the principle of molecular beacons. The method uses commercially-available reagents and allows quantitative monitoring of the 3'-proccessing in a multiwell plate-reader format.
Materials and methods
Materials The 3'-processing substrate (5'-[FAM]-ACT GCT AGA GAT TTT CCA CGT GGA AAA TCT CTA GCA GT-[DABCYL]-3') and a control substrate (5'-[FAM]-T GCT AGA GAT TTT CCA CGT GGA AAA TCT CTA GCA–[DABCYL]-3') were labeled at the 5' end with a fluorophore (carboxyfluorescein, FAM) and a quencher (4-[4'-dimethyl-aminophenylazo] benzoic acid, DABCYL) at the 3' end. These substrates were synthesized by Shanghai Sangon Bio Inc (Shanghai, China). Taq DNA polymerase and all restriction endonucleases used in these experiments were purchased from Dalian Takara Bio Inc (Dalian, China). Piperazine-N,N'-bis-2-ethanesulfonic acid (PIPES) was purchased from Roche Diagnostics (Shanghai, China). Nickel-chelating column (Chelating Sepharose Fast Flow) was purchased from Amersham Pharmacia Biotech (Piscataway, NJ, USA). N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic (HEPES), imidazole, and isopropyl-beta-D-thiogalactopyra-noside (IPTG) were obtained from Merck (Hohenbrunn, Germany). Of the 2 IN inhibitors, baicalein was purchased from Fluka (Buchs SG, Switzerland) and myricetin was purchased from Acros Organics (Morris Plains, NJ, USA). Tryptone and yeast extract were purchased from Oxoid Limited (Basing-stoke, Hampshire, UK). All other chemicals were purchased from Sigma Chemical Company (St Louis, MO, USA).
Protein expression and purification The cDNA of HIV-1 HXB2 (one of HIV-1 virus strains, GenBank accession number K03455) IN was used as a template, F185K/C280S site-directed mutagenesis was done by overlapping PCR to enhance the protein solubility as described by Jenkins et al[26]. The IN gene was modified to contain an Nde I site at the 5' end and a BamH I site and a termination codon (TAG) at the 3' end, then cloned into a pET28(a)+ vector to construct a pET-IN recombinant plasmid. The pET-IN was expressed in the Escheria coli BL21 strain (DE3) plysS cells as a N-terminal 6-histidine tag fusion protein. Cells were grown at 37 °C in super broth medium (16 g/L tryptone, 10 g/L yeast extract, and 5 g/L NaCl, pH 7.0) containing 50 mg/L kanamycin to 0.8 of the optical density (OD) for cells solution at 600 nm, and induced with 0.4 mmol/L IPTG for 4 h. The cells were harvested by centrifugation at 4200×g for 20 min at 4 °C and resuspended in buffer A (20 mmol/L HEPES, pH 7.4, 1 mol/L NaCl, 2 mmol/L β-mercaptoethanol, and 5 mmol/L imidazole) with 0.3 g/L lysozyme and sonicated in an ice bath until lysis was complete. The supernatant was collected by centrifugation, filtered, and applied to a Ni-affinity column equilibrated with buffer A. The column was washed first with 20 and 60 mmol/L imidazole in buffer A, and then with a linear gradient of 100–300 mmol/L imidazole in the same buffer. Fractions containing HIV IN were pooled together and dialyzed against buffer B [20 mmol/L HEPES, pH 7.4, 1 mol/L NaCl, 0.1 mmol/L EDTA, 1 mmol/L dithiothreitol, and 10% (w/v) glycerol] for 24 h at 4 °C with intermittent changes of buffer to remove imidazole and stored at -80 °C. The protein concentration was determined by the Bradford assay with bovine serum albumin (BSA) as a standard[27]. To determine the purity of the proteins, SDS-PAGE was performed with a 5%–12% (w/v) gradient of acrylamide and 0.2% (w/v) N, N-methylene-bisacrylamide. Molecular weight standards used were carbonic anhydrase from bovine erythrocytes (28 500), albumin from chicken eggs (45 000), albumin from bovine serum (66 200), and phosphorylase β from rabbit muscles (94 000).
3'-processing assay The 3'-processing assay was performed at 37 °C in 96-well plates (Corning Incorporated, NY, USA) in a final volume of 100 μL per well. The reaction mixture contained 25 mmol/L PIPES, pH 7.0, 10 mmol/L β-mercaptoethanol, 5% glycerol, 0.1 g/L BSA, 10 mmol/L MnCl2, and 50 mg/L purified IN. The reaction was initiated by the addition of 400 nmol/L of the 3'-processing substrate. The fluorescence signal was continuously monitored in a 1420 Victor Multilabel counter (Perkin Elmer, Boston, MA, USA) under the settings of 485 nm excitation and 535 nm emission. Enzyme-free control wells were set under the same reaction conditions without IN in the reaction mixture to monitor the background signal. Substrate control wells were set with all the reagents except for the substitution of the 3'-processing substrate by a control substrate. These 2 kinds of control wells were continuously observed as the 3'-processing reaction. All the reagents used in the assay were freshly made each time.
Inhibition of the integrase 3'-processing activity To determine the inhibition of the integrase 3'-processing, the reactions were carried out in the 100 μL total volume with 25 mmol/L PIPES, pH 7.0, 10 mmol/L β-mercaptoethanol, 5% glycerol, 0.1 g/L BSA, 10 mmol/L MnCl2, 50 mg/L purified IN, and various concentrations of inhibitors diluted in DMSO. The reactions were initiated by the addition of 400 nmol/L of the 3'-processing substrate. The reagents mixed with 10% DMSO, but no inhibitor was set as the drug-free control; the background control was set as mentioned in the 3'-processing assay. Rate data were collected at 37 °C in the 1420 Victor Multilabel counter. IC50 values were determined by a nonlinear regression curve fit of the rate data to the following equation:
Results
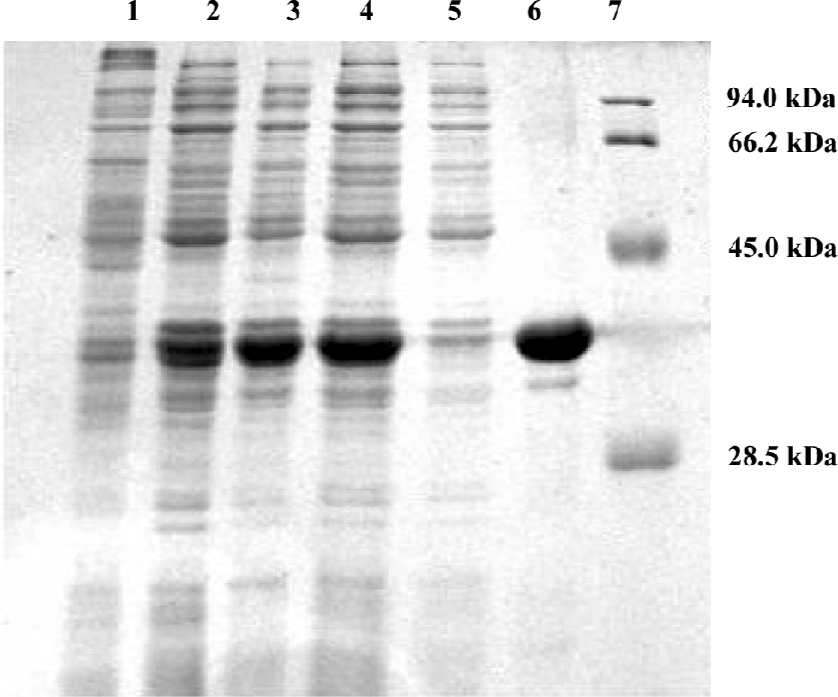
HIV IN expression and purification HIV-1 IN was expressed in the E coli BL21 (DE3) plysS strain and purified from the soluble supernatant using nickel affinity chromatography as described in the Material and methods section. The 12% SDS-PAGE used to separate the total protein of the cell lysate (lanes 1–3) indicated that after 4 h inducement, the expression of IN significantly exceeded the expression level of other cellular proteins (Figure 1). The molecular weight of purified IN was about 33 kDa, with a purity of about 95%.
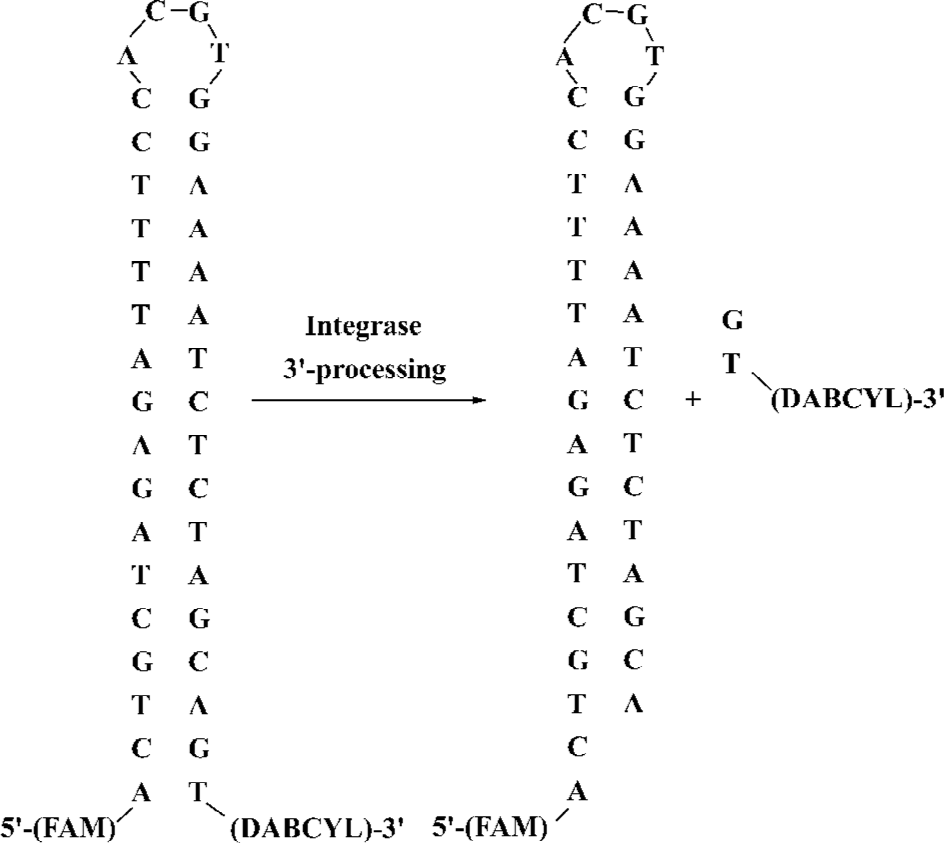
Principles of high-throughput real-time assay based on molecular beacons In the present study, a real-time assay for the integrase 3'-processing reaction in solution was proposed (Figure 2). It is an in vitro cell-free and virus-free system in which the recombinant HIV-1 IN is incubated with a 38 mer oligonucleotide substrate with a sequence identical to the U5 end of HIV-1 LTR. As part of the 3'-processing reaction, the terminal 3'-dinucleotide, containing the quencher, is cleaved from the oligonucleotide, and the quenching effect is abrogated to result in a marked increase in the fluorescence intensity of the reaction mixture. The rate of the increase in fluorescence intensity is proportional to the velocity of the 3'-processing reaction because the change in fluorescence intensity is dependent on the release of the quencher-containing dinucleotide. This assay is specific to the IN 3'-processing reaction.
Optimization of 3'-processing assay To optimize the robustness of this assay, we performed tests to investigate the effects of substrates, enzyme, and the metal ion concentrations on IN activity. In the assay, substrate concentrations ranged from 10 to 1000 nmol/L, the recombinant IN concentrations ranged from 10 to 100 mg/L, and the metal ion concentrations were chosen from 1 to 10 mmol/L. At the same time, the reference controls of the substrate and metal ion concentrations were set with all the reagents without IN. At substrate concentrations below 400 nmol/L, the fluorescence intensity grew in proportion to the increase in the substrate concentration. The fluorescence intensity of the reaction mixture grew slower than that of the control without IN at substrate concentrations exceeding 400 nmol/L. The assay showed that the reaction rate was also dependent on the protein concentration, and the optimal IN concentration was 50 mg/L. The fluorescence intensity was enhanced with increasing Mn2+ concentrations, but no obvious change was observed at Mn2+ concentrations greater than 10 mmol/L. Here, 10 mmol/L Mn2+ was chosen as an optimal concentration.
Under the optimal reaction conditions, a continuous and sharp fluorescence signal growth representing the accumulation of the 3'-processing product was observed in the first 2 h, but the growth rate slowed down and the real-time curve reached a plateau after that point in time (Figure 3A). The 3'-processing reaction under optimal conditions showed 560 000 cps of the fluorescence intensity during 3.5 h, while the enzyme-free control and the substrate control remained under 30 000 and 60 000 cps, respectively, during the entire reaction.
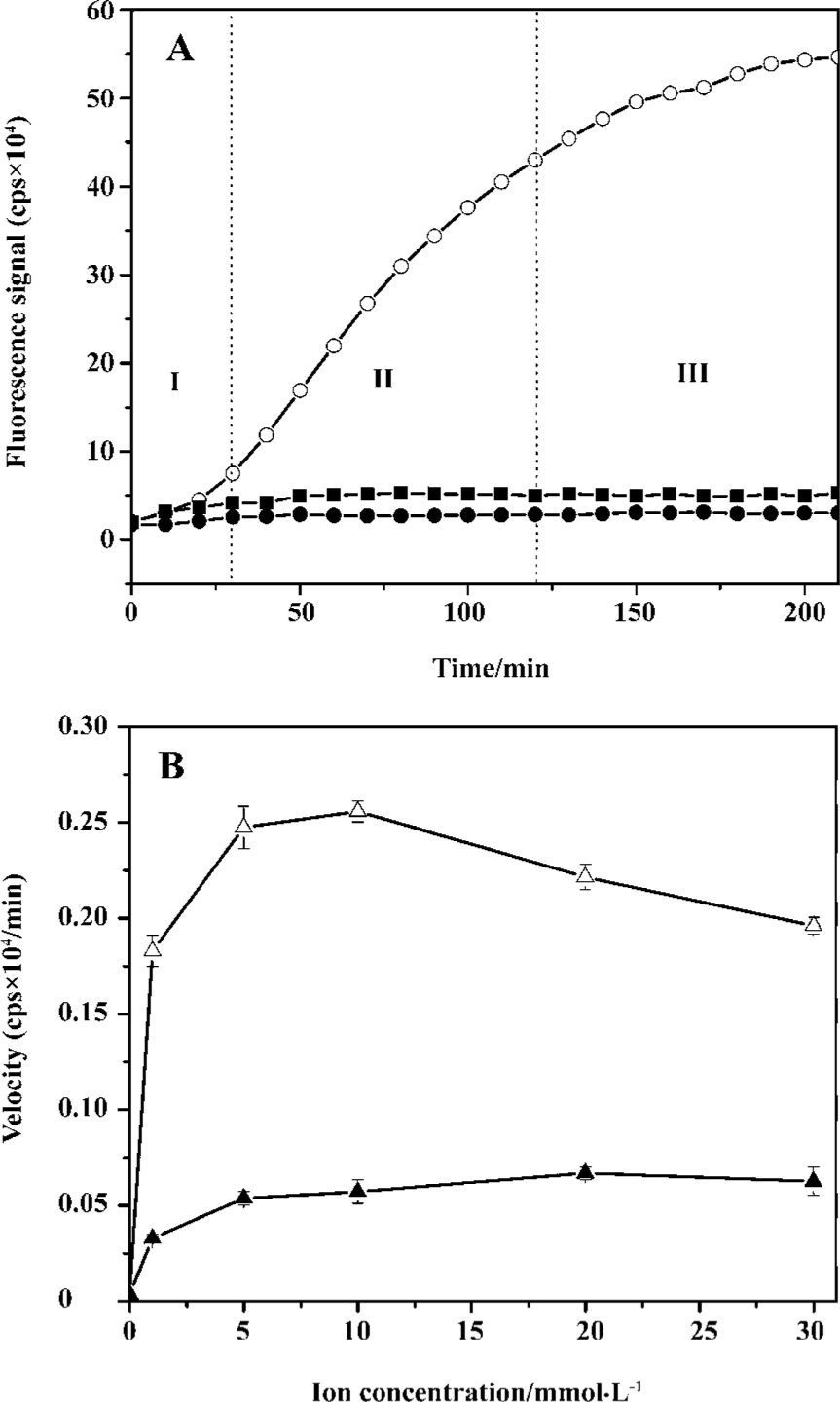
To demonstrate that the cleavage observed in the presence of the enzyme is really a 3'-processing activity, but not a non-specific hydrolysis, a control substrate was designed as an oligonucleotide without AC dinucleotide at the 5' end and GT dinucleotide at the 3' end. HIV-1 IN proved to be sensitive and specific to its substrate during the 3'-processing reaction. If this cleavage of 5'-fluorophore or 3'-quencher results from the 3'-processing reaction, no fluorescence signal increase can be observed for the reaction of IN with the control substrate. If not, fluorescence intensity would increase with time. In this assay, no notable fluorescence signal increase was observed for the substrate control wells, and the substrate control signal was approaching that of the enzyme-free control. Compared with the 2 controls, the fluorescence intensity increased significantly with the reaction time in the system of IN with the 3'-processing substrate (Figure 3A). It is clear that the increase of fluorescence signal in this assay is specifically due to the IN-mediated 3'-processing reaction.
This enzymatic assay for IN 3'-processing activity described here does not strictly correspond to Michaelis–Menten conditions as well as most of the enzymatic assays for IN activities[11–20]. The time-dependent fluorescence intensity for the 3'-processing reaction in this assay displayed 3 distinct phases (Figure 3A): the lag in phase I lasted about 20–30 min, followed by an apparent linear increase (phase II) for about 1.5 h, and an equilibrium phase (phase III) after 2 h. This phenomenon is similar to the observation made by Smolov et al[20]. This lag may be due to the slow binding of IN to DNA for about 20 min to form a stable DNA-IN complex[20,29] because the DNA substrate was added at the end to initiate the reactions and there was no pre-incubation of DNA with IN in our assay. The DNA-binding step can be measured by steady-state fluorescence anisotropy [20,28–30], which is based on the principle of depolarization of light by a fluorescent probe caused by the rotational diffusion that occurs between excitation and emission. IN-bound DNA gave a higher anisotropy value than free DNA, and the equilibrium reached after a 20 min incubation of IN with the DNA substrate[20]. Furthermore, phase I was reduced when IN was pre-incubated with DNA for 10–20 min at 37 °C prior to the addition of the metal ion cofactor, and pre-incubation did not significantly influence the product formation in phases II and III.
The effects of Mn2+and Mg2+ on the IN 3'-processing activity in this assay were compared (Figure 3B). Mg2+ is believed to be relevant for viral replication in vivo, but Mn2+ is frequently used in most in vitro assays instead of Mg2+ because higher activity of HIV-1 IN is observed with Mn2+. The optimum concentration of Mg2+ is 20 mmol/L, while the optimum concentration of Mn2+ is 10 mmol/L. The Mn2+-dependent activity is about 4 times that of Mg2+-dependent activity and is similar to the observation by Tramontano et al[31].
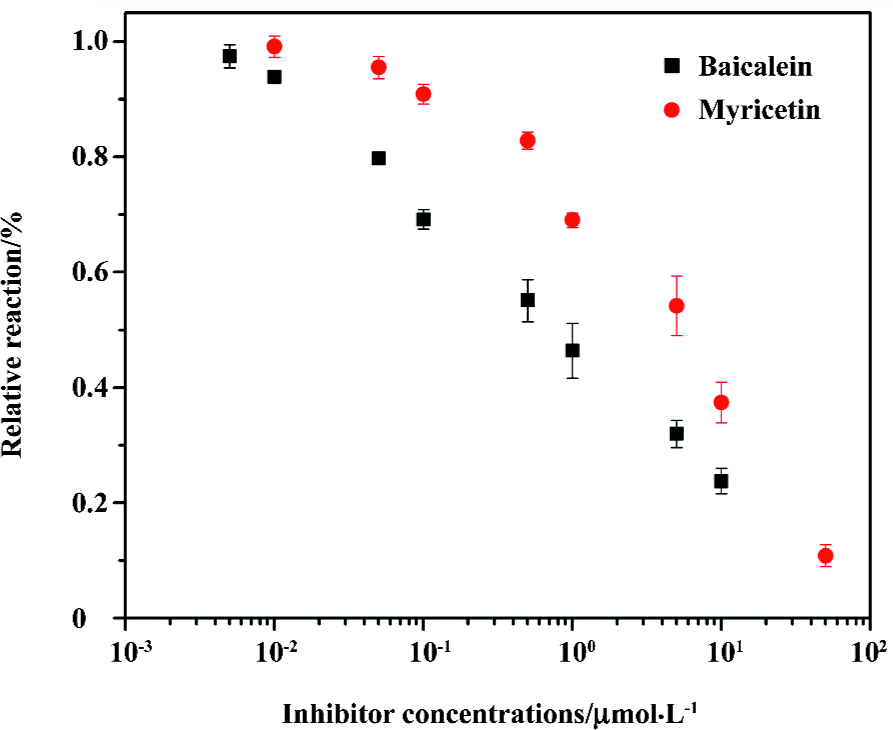
Integrase inhibitor screening Baicalein and myricetin, reported to be HIV-1 IN 3'-processing inhibitors[31], were employed to test the performance of this assay in IN inhibitor screening (Figure 4). The IC50 values of baicalein and myricetin obtained in our assay are 0.89 µmol/L and 5.6 µmol/L, respectively, which are similar to the values (0.7 µmol/L for baicalein and 7.0 µmol/L for myricetin) obtained from a radiolabeled substrate assay by Tramontano et al[31]. This shows that the assay can be used for the identification of HIV-1 IN 3'-processing inhibitors.
Discussion
In the present work, we propose a high-throughput real-time assay for the 3'-processing activity of IN. Based on the principle of molecular beacons, the assay has significant advantages. First, since no precoating of the assay plates with enzyme substrate or capture reagents was required, the measurements of the homogeneous assay yielded highly reproducible data with enzyme reactions performed in solution and no loss of enzyme activity. Second, the identification of rate-limiting steps in the overall catalytic process is important, especially for pharmacological purposes. The assay has high sensitivity and real-time monitoring, and it can be used for the kinetic study of the IN 3'-processing reaction. Third, compared to current assays, this assay can be used for different purposes because it can be conducted in microplates, microtubes, or other reaction containers, and the results can be monitored by any fluorescence detecting instruments. To test the performance of this assay in antiviral compounds screening, 2 reported 3'-processing inhibitors were used as examples. The IC50 values of baicalein and myricetin observed in our assay were similar to the previous experiment data[31]. In summary, the assay presented here can be used both for monitoring the HIV-1 IN 3'-processing activity and for inhibitor screening.
Acknowledgements
We thank Dr Xue-mei MA, Mr Chuan-chen LU, and Mr Fei XIE of Beijing University of Technology for their helpful discussion for the fluorescence intensity measurements.
References
- Engelman A, Mizuuchi K, Craigie R. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 1991;67:1211-21.
- Pluymers W, De Clercq E, Debyser Z. HIV-1 integration as a target for antiretroviral therapy: a review. Curr Drug Targets Infect Disord 2001;1:133-49.
- Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002;110:521-9.
- Brown PO. Integration. In: Coffin JC, Hughes SH, Varmus HE, editors. Retroviruses. New York: Cold Spring Harbor Lab Press; 1999. p 161–203.
- Asante-Appiah E, Skalka AM. HIV-1 integrase: structural organization, conformational changes, and catalysis. Adv Virus Res 1999;52:351-69.
- Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 1994;266:1981-6.
- Goldgur Y, Craigie R, Cohen GH, Fujiwara T, Yoshinaga T, Fujishita T, et al. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc Natl Acad Sci USA 1999;96:13040-3.
- Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, et al. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc Natl Acad Sci USA 2000;97:8233-8.
- Pluymers W, De Clercq E, Debyser Z. HIV-1 integration as a target for antiretroviral therapy: a review. Curr Drug Targets Infect Disord 2001;1:133-49.
- Esposito D, Craigie R. HIV integrase structure and function. Adv Virus Res 1999;52:319-33.
- Marchand C, Neamati N, Pommier Y. In vitro human immunodeficiency virus type 1 integrase assays. Methods Enzymol 2001;340:624-33.
- Chow SA. In vitro assays for activities of retroviral integrase. Methods 1997;12:306-17.
- Mazumder A, Pommier Y. Processing of deoxyuridine mismatches and abasic sites by human immunodeficiency virus type-1 integrase. Nucleic Acids Res 1995;23:2865-71.
- Craigie R, Mizuuchi K, Bushman FD, Engelman A. A rapid in vitro assay for HIV DNA integration. Nucleic Acids Res 1991;19:2729-34.
- Hazuda DJ, Hastings JC, Wolfe AL, Emini EA. A novel assay for the DNA strand-transfer reaction of HIV-1 integrase. Nucleic Acids Res 1994;22:1121-2.
- John S, Fletcher TM III, Jonsson CB. Development and application of a high-throughput screening assay for HIV-1 integrase enzyme activities. J Biomol Screen 2005;10:606-14.
- Vink C, Banks M, Bethell R, Plasterk RH. A high-throughput, non-radioactive microtiter plate assay for activity of the human immunodeficiency virus integrase protein. Nucleic Acids Res 1994;22:2176-7.
- Hawkins ME, Pfleiderer W, Mazumder A, Pommier YG, Balis FM. Incorporation of a fluorescent guanosine analog into oligonucleotides and its application to a real time assay for the HIV-1 integrase 3'-processing reaction. Nucleic Acids Res 1995;23:2872-80.
- Wang Y, Klock H, Yin H, Wolff K, Bieza K, Niswonger K, et al. Homogeneous high-throughput screening assays for HIV-1 integrase 3'-processing and strand transfer activities. J Biomol Screen 2005;10:456-62.
- Smolov M, Gottikh M, Tashlitskii V, Korolev S, Demidyuk I, Brochon JC, et al. Kinetic study of the HIV-1 DNA 3'-end processing single-turnover property of integrase. FEBS J 2006;273:1137-51.
- Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol 1996;14:303-8.
- Tan W, Wang K, Drake T. Molecular beacons. Curr Opin Chem Biol 2004;8:547-53.
- Li JJ, Fang X, Schuster SM, Tan W. Molecular beacons: a novel approach to detect protein-DNA interactions. Angew Chem Int Ed Engl 2000;39:1049-52.
- Tang Z, Wang K, Tan W, Li J, Liu L, Guo Q, et al. Real-time monitoring the nucleic acids ligation in homogenous solution using molecular beacon. Nucleic Acids Res 2003;31:e148.
- Biggins JB, Prudent JR, Marshall DJ, Ruppen M, Thorson JS. A continuous assay for DNA cleavage: The application of “break lights” to enediynes, ion-dependent agents, and nucleases. Proc Natl Acad Sci USA 2000; 97: 13 537–42.
- Jenkins TM, Engelman A, Ghirlando R, Craigie R. A soluble active mutant of HIV-1 integrase: involvement of both the core and carboxyl-terminal domains in multimerization. J Biol Chem 1996;271:7712-8.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248-54.
- Agapkina J, Smolov M, Barbe S, Zubin E, Zatsepin T, Deprez E, . Probing of HIV-1 integrase-DNA interaction using novel analogs of viral DNA. J Biol Chem 2006; 281: 11 530–40.
- Pinskaya M, Romanova E, Volkov E, Deprez E, Leh H, Brochon JC, et al. HIV-I integrase complexes with DNA dissociate in the presence of short oligonucleotides conjugated to acridine. Biochemistry 2004;43:8735-43.
- Deprez E, Tauc P, Leh H, Mouscadet JF, Auclair C, Hawkins ME, et al. DNA binding induces dissociation of the multimeric form of HIV-1 integrase: a time-resolved fluorescence anisotropy study. Proc Natl Acad Sci USA 2001;98:10090-5.
- Tramontano E, Onidi L, Esposito F, Badas R, Colla PL. The use of a new in vitro reaction substrate reproducing both U3 and U5 regions of the HIV-1 3'-end increases the correlation between the in vitro and in vivo effects of the HIV-1 integrase inhibitors. Biochem Pharmacol 2004;67:1751-61.