
Biphasic effects of haloperidol on sodium currents in guinea pig ventricular myocytes
Introduction
Haloperidol is used to treat certain mental/mood disorders (eg schizophrenia and schizoaffective disorders). In 2004, it was one of the 6 most frequently prescribed antipsychotic drugs[1]. However, changes in the electrokardiogram (EKG) pattern (Q-T prolongation[2−4], T wave inversion, and ST segment depression) after haloperidol treatment are common. Excessive prolongation in the Q-T interval may lead to after-depolarization which may result in torsades de points (Tdp)[5]. It was demonstrated that haloperidol decreased inward sodium currents (INa) in a frequency-dependent manner in cardiac myocytes[6]. Yet another recent investigation revealed that haloperidol had biphasic frequency-dependency on cardiac myocytes, frequency-dependent decrease in intraventricular conduction, and reverse frequency-dependent prolongation of repolariza-tion[7]. The aim of the present study was to examine the effect of haloperidol on INa in isolated guinea pig ventricular myocytes in order to explore the possible mechanism(s) of drug-induced arrhythmias.
Materials and methods
Myocyte preparation and solutions Guinea pig ventricular myocytes were enzymatically dissociated as described previously[8]. In brief, guinea pigs of either gender (300±20 g) were anesthetized, and the hearts were quickly removed and placed in oxygenated Tyrode’s solution. The hearts were mounted on a Langendorff apparatus and perfused with 37 °C oxygenated Tyrode’s solution containing (in mmol/L): 140 NaCl, 5.4 KCl, 1 MgCl2, 2 CaCl2, 0.33 NaH2PO4, 10 glucose, and 10 HEPES (pH adjusted to 7.4 with NaOH). Then after perfusion with Ca2+-free Tyrode’s solution for 5–10 min, the hearts were digested with solution containing 0.5 mg/mL collagenase (type II, Worthington, Lakewood, New Jersey, USA) and 1 mg/mL bovine serum albumin (BSA) (Sigma, Saint Louis, Missouri, USA). The isolated myocytes were stored in high-K+ media containing (in mmol/L): 10 KCl, 10 KH2PO4, 120 potassium glutamate, 10 taurine, 1.0 MgSO4, 20 glucose, and 0.5 EGTA (pH adjusted to 7.2 with KOH) and were used within 8 h of isolation.
The myocytes were placed in a chamber (about 0.3 mL) on an inverted microscope and superfused with solution containing (in mmol/L): 5 NaCl, 135 CsCl, 1 MgCl2, 10 glucose, 0.5 CaCl2, 0.5 CoCl2, and 5 HEPES (pH adjusted to 7.4 with CsOH) at 21–22 °C. Only quiescent, rod-shaped cells showing clear striations were selected for the experiments.
Electrophysiological recording The INa was recorded by whole cell patch-clamp technique (HEAK EPC-10 plus, HEAK Instruments, D-67466 Lambrecht/Pfalz, Germany). Data were acquired at 50 kHz, and command pulses were generated by PULSE+PULSEFIT (HEAK, D-67466 Lambrecht/Pfalz, Germany). Recordings were low-pass filtered at 5 kHz with an 8-pole Bessel filter (HEAK Instruments, D-67466 Lambrecht/Pfalz, Germany) and stored on hard disk.
Borosilicate glass [1.5 mm outside diameter (OD)] patch pipettes were filled with (in mmol/L) 5 NaCl, 20 CsCl, 110 CsF, 1 MgCl2, 5 HEPES, 5 EGTA, and 5 Mg2ATP (pH adjusted to 7.2 with CsOH). Junction potentials were compensated before the pipette touched the cell. Series resistance (Rs) and capacitance were electronically compensated; Rs=1.1±0.2 mV (n=30) after compensation. Caution was taken to ensure that the voltage drop across the Rs was <5 mV. Data were discarded if there was evidence of inadequate voltage control.
Statistical analysis A nonlinear curve fitting was done in Clampfit 9.0 (Axon, Sunnyvale, California, USA) and Sigmaplot 8.0 (SPSS, Chicago, Illinois, USA). Paired and unpaired Student’s t-tests were used to evaluate differences between 2 means. P<0.05 was considered to indicate significance. Group data are expressed as mean±SEM.
Results
Inhibitory effect of 0.3 µmol/L haloperidol on INa
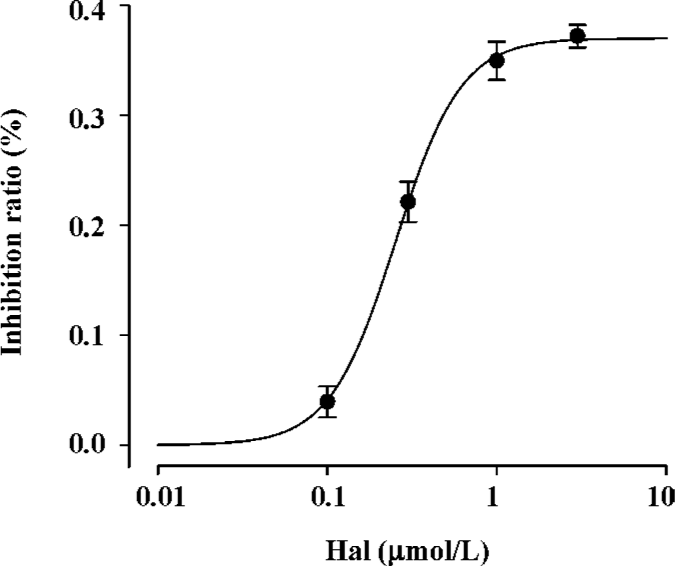
Concentration-dependent effect of haloperidol (0.1-3.0 µmol/L) on INa Under our current experimental condition, the maximum activation voltage for INa was between -30 and -40 mV. Haloperidol (0.1−3.0 µmol/L) elicited a concentration-dependent inhibition on INa (Figure 1). INa elicited by depolarization to -35 mV in haloperidol was normalized by control INa in the same cell. The inhibition ratios of 0.1, 0.3, 1, and 3 μmol/L haloperidol were 3.94%±0.014% (n=6, P<0.05), 22.14%±0.02% (n=7, P<0.001), 34.97%±0.02% (n=6, P<0.001), and 37.21%±0.10% (n=6, P<0.001), respectively. If administered higher concentrations (10 µmol/L), there was no current appearing even under about -35 mV, which supposed the cell would be dead. So it was supposed that 3 µmol/L haloperidol was the concentration approached the maximum effect. Using the inhibition ratio to plot against haloperidol concentration, the concentration-response curve was fitted by the Hill formulation {y=1/[1+(C/IC50)b]}. On the basis of cell-by-cell fits, the haloperidol concentration giving a 50% block (IC50) at -35 mV was 0.253±0.015 µmol/L with a Hill coefficient of 2.24 (n=25 cells).
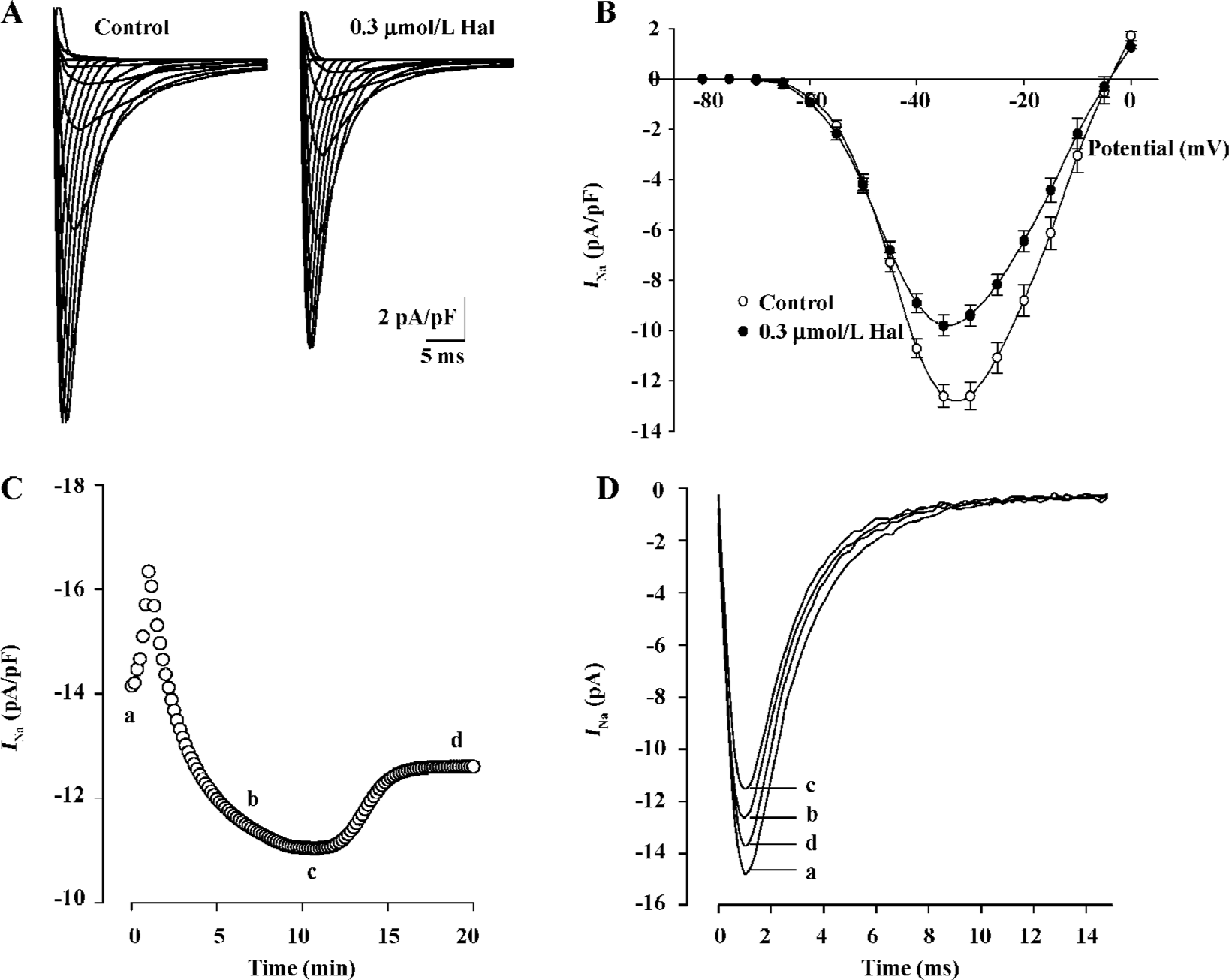
Effect of 0.3 µmol/L haloperidol on INa As IC50 (0.253 µmol/L) was near 0.3 µmol/L, we made 0.3 µmol/L as representative concentration to investigate the changes of various parameters of inhibition effect on INa by the drug. Families of capacity- and leak-corrected INa elicited with 40 ms time courses from -130 mV to between -80 and +15 mV of the control and 0.3 µmol/L haloperidol are shown in Figure 2A. Inward currents were substantially reduced by 0.3 µmol/L haloperidol. Taking the test voltage as the X axis and current density (pA/pF) as the Y-axis, current-voltage (I–V) relationship curves are plotted in Figure 2B. The time course of block of INa by haloperidol at -35 mV and recovery on washout are illustrated in Figure 2C. The block reached a steady state after about 10 min, but recovery was biphasic and incomplete during the 10 min washout period. The block of peak INa was associated with the slowing of activation and inactivation kinetics. In superimposed traces (Figure 2D), peak times had nearly no changes which reflected no changes of activation kinetics, but inactivation courses were earlier than the control.
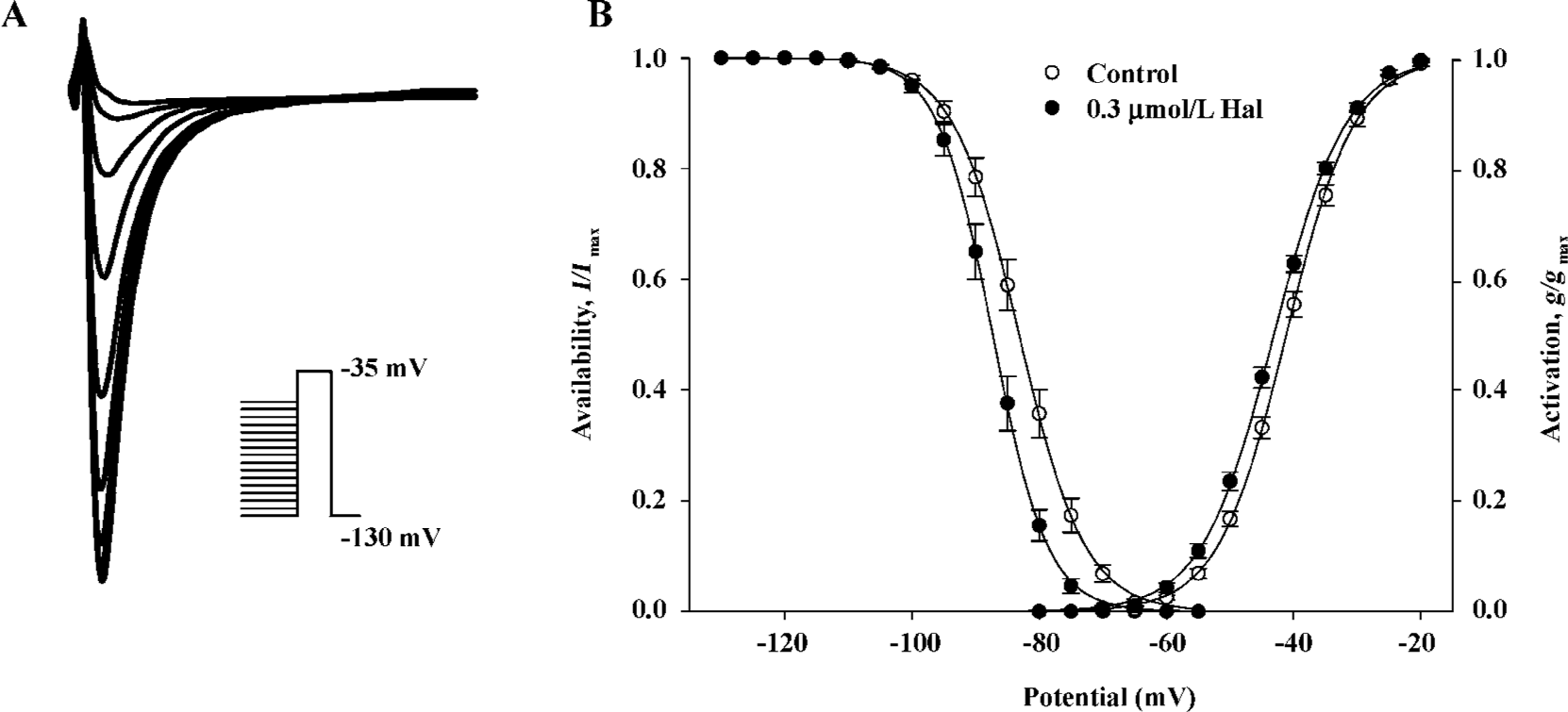
Voltage dependence of conductance and availability of INa before and after 0.3 µmol/L haloperidol treatment The voltage dependence of the conductance activation variable (g/gmax) of INa was determined from I–V relationships for each cell (Figure 3B) and conductance equation: g=INa/(Vm–Vr) (where INa is current density, Vm and Vr are membrane potential and reverse potential, respectively) and was fitted to the Boltzmann equation: y=1/{1+exp[(Vm–V0.5)/S]} to obtain the voltage for half-activation (V0.5) and slope factor (S). The voltage dependence of availability (I/Imax) was determined as illustrated in Figure 3A and was also fitted to the Boltzmann equation. Figure 3B shows that 0.3 μmol/L haloperidol shifted the midpoint for conductance and availability of INa to more negative potentials. The V0.5 for activation shifted 2.09 mV, from -41.21±0.50 mV in the control to -43.86±0.47 mV in haloperidol (n=7, P<0.01). The shift of the availability curve was slightly more, 4.09 mV, from -86.11±0.98 mV in the control to -90.20±1.04 mV in haloperidol (n=7, P<0.001). The S values for conductance, which were not significantly altered, were -5.30±0.14 and -5.06±0.18 mV (n=7, not significant) and in contrast, the S values for availability were 5.00±0.11 and 4.67±0.18 mV (n=7, P<0.05) in the control and haloperidol, respectively.
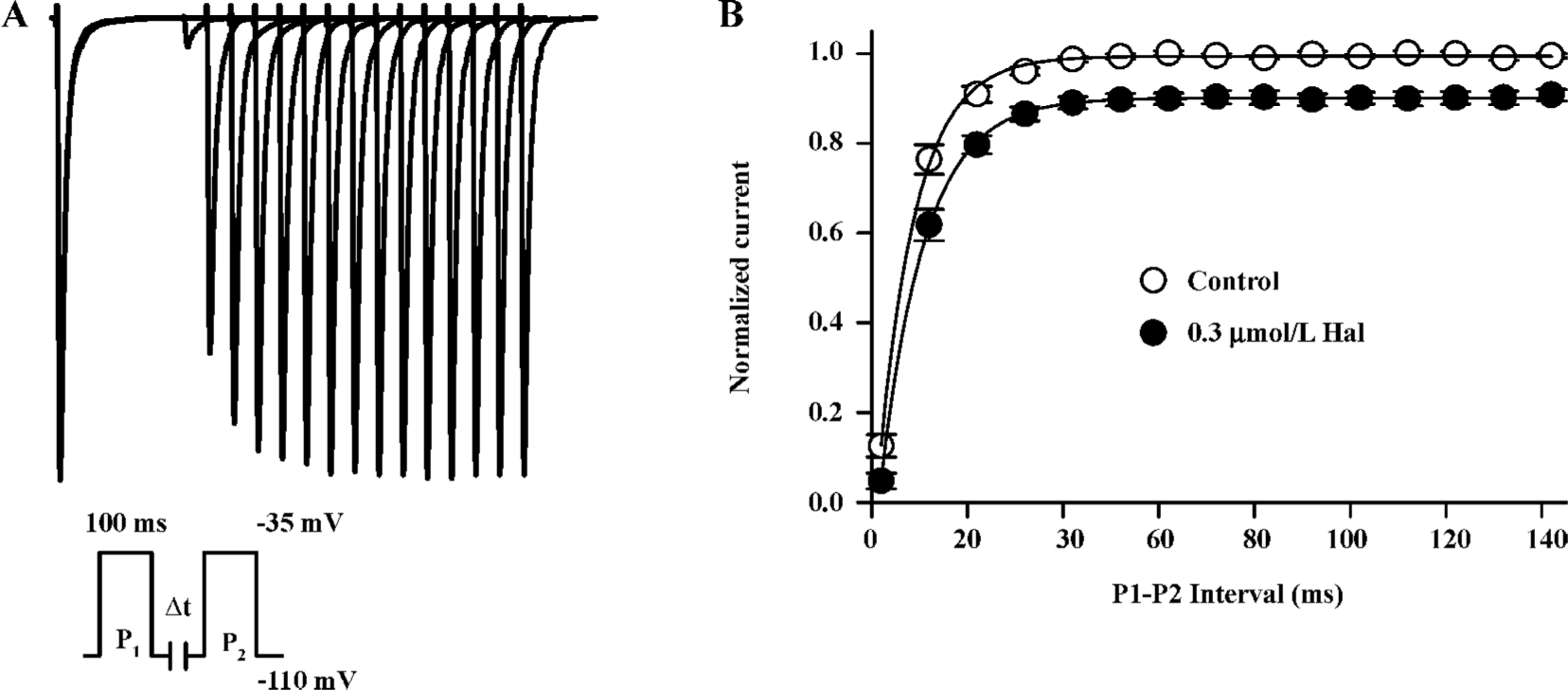
Kinetics of recovery from inactivation The recovery of INa from inactivation was studied with a paired-pulse protocol. Superimposed currents and the time course of recovery are illustrated in Figure 4. The INa recovery was not complete (recovery was about 90.14%±1.4%, n=6) and well fitted by monoexponential functions with time constants of 7.84±0.74 ms in the control and 9.20±0.71 ms in 0.3 µmol/L haloperidol (n=6, P<0.01). This indicates that 0.3 µmol/L haloperidol did affect the recovery of INa from inactivation at hyperpolarized potentials.
Biphasic effect of 0.03 µmol/L haloperidol on INa
Enhanced effect of 0.03 µmol/L haloperidol on INa Under lower concentrations of haloperidol (0.01 and 0.03 µmol/L), INa appeared enhanced effect. The effect was enhanced more obviously in 0.03 µmol/L (by 57.01%±6.35%, n=6, P<0.01). Initial current-voltage superimposed curves before and after 0.03 µmol/L haloperidol treatment are shown in Figure 5A, and the I–V curves are indicated in Figure 5B.
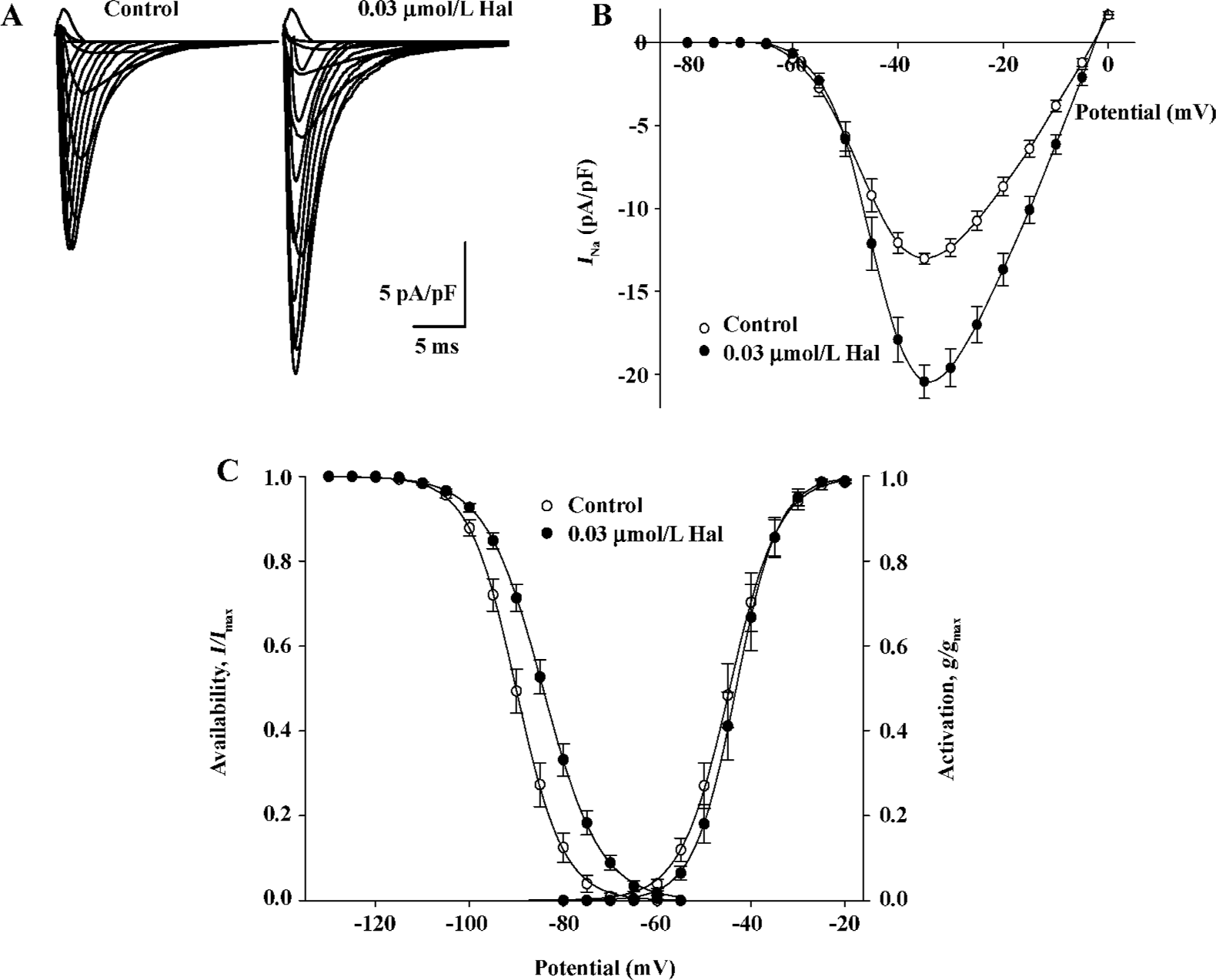
Voltage dependence of conductance and availability of INa by 0.03 µmol/L haloperidol The midpoint for conductance (g/gmax) and availability (I/Imax) of INa was shifted to positive potentials by 0.03 µmol/L haloperidol (Figure 5C), in which V0.5 for activation increased by 1.38 mV from -44.52± 1.56 mV in the control to -43.15±1.48 mV in 0.03 µmol/L haloperidol (n=6, P<0.01); S values were -4.86±0.20 mV in the control and -4.09±0.16 mV in haloperidol (n=6, P<0.01), respectively. The V0.5 for inactivation increased by 5.69 mV from -90.02±1.15 to -84.33±1.01 mV (n= 6, P<0.01); the S value for inactivation was enhanced from 4.87±0.19 to 6.01±0.21 mV before and after 0.03 μmol/L haloperidol (n=6, P<0.01).
Discussion
INa is the major depolarizing current of the working cardiac myocytes. Class I anti-arrhythmic drugs exert their actions by inhibiting INa in cardiac myocytes. Animal studies have found that haloperidol depresses action potential amplitude and phase 0 maximum upstroke velocity (Vmax), in which the effects are similar to that of quinidine.
Our study showed that haloperidol (0.1−3 µmol/L) depressed INa in a concentration-dependant manner (Figure 1), and the results are in accordance with previous studies that show that haloperidol has the same effects on the action potentials of isolated Purkinje fiber and papillary muscles.
A negative shift in the activation/inactivation curves were observed after 0.3 µmol/L haloperidol, where the voltage at both half activation and half inactivation was decreased. We assumed haloperidol caused an early activation and inactivation of sodium channels. According to the modulated receptor theory, the drug mainly affected the inactivated state of sodium channels. A slow recovery of the inactivation curve indicated recovery of sodium channels after inactivation was slow, which is a reflection on the slow unbinding rate of the dissociation of the drug from the channel receptor. After administering 0.3 µmol/L haloperidol, the numbers of sodium channels recovered by 90.14%±1.4% (n=6) and did not completely recover with longer time intervals. This might be because only about 90% of the channels were dissociated after giving 0.3 µmol/L haloperidol and this dissociation rate remained constant as the time for haloperidol exposure was prolonged. Haloperidol delayed the recovery of INa from inactivation and this explains the negative shift in the inactivation curve[9]. We conclude that haloperidol may have some properties of class I anti-arrhythmic drugs[10].
Paradoxically, haloperidol at lower concentrations (0.01 and 0.03 µmol/L) stimulated INa. Haloperidol (0.03 µmol/L) caused a positive shift in the inactivation curve and enhanced the kinetic parameters of the sodium channel (Figure 5). We found no similar results in the literature and to our knowledge, report on this biphasic effect of haloperidol for the first time.
The inhibition of sodium channels by haloperidol decreases conductivity and makes the heart more susceptible to the development of unidirectional conduction block. A depressed membrane potential not only decreases Vmax, but also inhibits further propagation of the impulse and results in re-entrant excitation. However, pathogenesis of fatal arrhythmia is complex. Experimental models of Tdp are usually associated with the induction of early after-depolarization (EAD) and there is a participation of a re-entry mechanism. The ionic basis of EAD is related to activated L-type Ca2+ currents during plateau, increased sodium influx, and the inhibition of K+ efflux via delayed rectified potassium channels. Therefore, we deduce that the stimulation of INa by low concentrations of haloperidol contributes to the formation of EAD and this may be one of the mechanisms of haloperidol-induced Tdp.
Arrhythmia is associated with both abnormalities in depolarization and repolarization processes. The ionic currents which participate in the repolarization are even more complicated. As part of the investigation, we will conduct further research on haloperidol’s effect on other ionic channels as well as their subtypes to elucidate the electrophysiological mechanisms of haloperidol-induced arrhythmia; this may add weight for understanding drug-induced arrhythmias.
Acknowledgements
This research would not have been possible without the technical support of Prof Man-wen JIN from the Department of Pharmacology, Tongji Medical University and Prof Gui-rong LI from the Internal Medicine Division, University of Hong Kong.
References
- Si TM, Shu L, Yu X, Ma C, Wang GH, Bai PS, et al. Antipsychotic drug patterns of schizophrenia in China: a cross-sectioned study. Chin J Psychiatry 2004;37:152-5.
- Gury C, Canceil O, Iaria P. Antipsychotic drugs and cardiovascular safety: current studies of prolonged Q-T interval and risk of ventricular arrhythmia. Encephale 2000;26:62-72.
- Zhou GB. Observation of electrocardiogram on patients taking haloperidol. Heilongjiang Med J 2002;25:95.
- Wu FM, Zhang JF. The effects of antipsychotic drugs on ECG of children. Chin J Psychiatry 1994;27:16.
- Gardner DM, Baldessarini RJ, Waraich P. Modern antipsychotic drugs: a critical overview. CMAJ 2005;172:1703-11.
- Ogata N, Narahashi T. Block of sodium channels by psychotropic drugs in single guinea-pig cardiac myocytes. Br J Pharmacol 1989;97:905-13.
- Mortl D, Agneter E, Krivanek P, Koppatz K, Todt H. Dual rate-dependent cardiac electrophysiologic effects of haloperidol: slowing of intraventricular conduction and lengthening of repolariza-tion. J Cardiovasc Pharmacol 2003;41:870-9.
- Li GR, Baumgarten CM. Modulation of cardiac Na+ current by gadolinium, a blocker of stretch-induced arrhythmias. Am J Physiol Heart Circ Physiol 2001;280:272-9.
- Bean BP, Cohen CJ, Tsien RW. Lidocaine block of cardiac sodium channels. J Gen Physiol 1983;81:613-42.
- Cambell TJ. Kinetics of onset of rate-dependent effects of class I antiarrhythmic drugs are important in determining their effects on refractoriness in guinea pig ventricle and provide a theoretical basis for their subclassification. Cardiovasc Res 1983;17:344-52.