
Minocycline inhibits 5-lipoxygenase activation and brain inflammation after focal cerebral ischemia in rats1
Introduction
Cerebral ischemia evokes secondary inflammation in the brain that contributes to ischemic insults[1]. In the delayed progression of ischemic stroke, postischemic inflammation may play an important role in brain damage[2]. As one of the pro-inflammatory molecules, 5-lipoxygenase (5-LOX) is the rate-limiting enzyme in the metabolism of arachidonic acid to produce leukotrienes, including leukotriene B4 (LTB4) and cysteinyl leukotrienes [CysLTs, namely leukotriene C4 (LTC4), LTD4 and LTE4]. The importance of 5-LOX in stroke has been proven in reports where the gene encoding the 5-LOX activating protein confers the risk of stroke[3,4]. Experimental studies have shown that the expression and the metabolite production of 5-LOX in the brain are increased after cerebral ischemia, and 5-LOX inhibitors exert neuroprotective effects on cerebral ischemic injury[5–10].
On the other hand, minocycline, a semisynthetic tetracycline antibiotic, has been reported to possess neuroprotective effects on cerebral ischemic injury[11–15] and other brain injuries[16–18]. The neuroprotective effect of minocycline relates to its anti-inflammatory and anti-apoptotic activities, such as inhibiting the activation and proliferation of microglia, the expressions of inducible nitric oxide synthase (iNOS), interleukin-1β converting enzyme and cyclooxygenase-2[11,12], and the caspase-dependent and independent cell apoptotic pathways[16–18]. However, the anti-inflammatory mechanisms of minocycline are not fully understood.
Recently, we reported that minocycline protected PC12 cells against in vitro ischemic-like injury or N-methyl-D-aspartate (NMDA)-induced excitotoxicity, and it could inhibit 5-LOX translocation to the nuclear membranes (a phenomenon of 5-LOX activation)[19,20]. These findings indicate that the in vitro protective effects of minocycline on ischemic or excitotoxic injuries may be partly mediated by inhibiting 5-LOX activation. In the present study, we determined whether minocycline exerts an in vivo anti-inflammatory effect that is mediated by inhibiting 5-LOX activation after focal cerebral ischemia in rats.
Materials and methods
Measurements of physiological variables Male Sprague-Dawley rats weighing 250–300 g (Experimental Animal Center, Zhejiang Academy of Medical Sciences, Hangzhou, China) were used in this study. The animals were housed under a controlled temperature (22±2 °C), 12 h light/dark cycle, and allowed free access to food and water. All experiments were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
The rats were anesthetized with chloral hydrate (400 mg/kg, ip). A polyethylene tube was inserted into the right femoral artery for continuously monitoring the blood pressure using a computer-assisted system (MedLab-U/4cs, Nanjing MedEase, Nanjing, China), and for measuring PaO2, PaCO2, and arterial blood pH (Blood Gas Analyzer ABL 330, Leidu, Copenhagen, Denmark). Blood glucose was monitored by the One Touch Basic Blood Glucose Monitoring System (Lifescan, Los Angeles, CA, USA). The rectal (core) temperature was measured and maintained at 37.0±0.5 °C with a heating pad and a heating lamp during the surgery. Percent changes in the regional cerebral blood flow (rCBF) over the middle cerebral arterial (MCA) territory (2 mm in diameter; 6 mm lateral and 2 mm caudal to bregma) were recorded as described[21] using a laser Doppler flowmeter (ML191, AD Instruments, Bella Vista, New South Wales, Australia), and the steady baseline value of rCBF before ischemia was 100%.
Middle cerebral artery occlusion Transient focal cerebral ischemia was induced by the suture occlusion method as previously described[22]. Briefly, after anesthesia, a midline incision was made in the neck, the right external carotid artery (ECA) and the right internal carotid artery (ICA) were carefully exposed and dissected, and a 3–0 (0.26 mm diameter) monofilament nylon suture was inserted from the ECA into the ICA to occlude the origin of the right MCA. After occlusion for 30 min, the suture was withdrawn to allow reperfusion, the ECA was ligated, and the incision was closed. The sham-operated rats underwent identical surgery, except that the intraluminal filament was not inserted. The achievement of the middle cerebral artery occlusion (MCAO) was confirmed by a reduction of 50% or more in the rCBF from the baseline value[23]. After surgery, the rats were kept for about 2 h in a warm box heated by lamps to maintain the body temperature.
Minocycline (Syowa Hakko, Tokyo, Japan) dissolved in sterile saline (22.5 and 45 mg/kg) was ip injected at 0.5 and 2 h after reperfusion on the first day, and twice daily on the second and third days. This dosage regimen was based on previous reports in studies of rat global and focal cerebral ischemia[11,12]. Equal volume (1 mL/kg) of saline was ip injected as the control.
Behavioral assessments Neurological deficit scores were evaluated 72 h after reperfusion according to the described method[24]; 0, no deficit; 1, flexion of contralateral forelimb upon lifting of the whole animal by the tail; 2, decrease of thrust toward contralateral plane; and 3, circling to the contralateral side. An inclined board test was performed to assess balance and coordination [25] based on the method developed by Yonemori et al[26]. The rats were placed on a board (50 cm×30 cm). Once they were stable, the board was inclined horizontally to vertically. The degree at which the animal fell from the board (holding angle) was recorded. The test was repeated 3 times and the average degree was used. All the behavioral and morphological changes were observed by the investigators who were blind to the treatments.
Histological examination After the behavioral assessments, the rats were anesthetized 72 h after reperfusion, and perfused transcardially with 4% paraformaldehyde after a saline prewash. The brains were removed and postfixed in 4% paraformaldehyde overnight, and transferred to 30% sucrose for 3 d. Six serial coronal slices were cut at 2 mm intervals from the frontal pole. Then, 2 sets of coronal sections (10 and 20 µm) were cut by cryomicrotomy (CM1900, Leica, Wiesbaden, Germany) from the slices. After being stained with 1% toluidine blue, the 20 µm thick sections were used for gross photographic examination, while the 10 µm sections were used for microphotographic or immunohistochemical examination.
In the gross photographs, the lesion area of the brain tissue was defined as an area with reduced Nissl staining, and confirmed by light microscopy to have dark pyknotic-necrotic cell bodies. The lesion areas were determined using an image analysis program (AnalyPower1.0, Zhejiang University, Hangzhou, China). The lesion volume of each section was calculated as: lesion area×slice thickness (2 mm), and the total lesion volume was the summation of the lesion volumes of all sections. In the microphotographs, the neurons in the temporoparietal cortex III and IV layers adjacent to infarcted area (0.2–0.4 mm caudal to bregma) were immunostained with a mouse monoclonal antibody against neuronal nuclei (NeuN) as described later, and neuron density was counted. The neurons or immunostained cells were randomly counted in three 200 µm2 squares at the upper, middle, and lower sites of the boundary zone adjacent to the ischemic core, and then were averaged.
Immunohistochemical analyses In the 10 µm sections, endogenous peroxidase activity was eliminated by reaction with 3% hydrogen peroxide for 30 min, and non-specific binding of IgG was blocked by incubation with 5% normal goat serum for 2 h at room temperature. The brain sections reacted overnight at 4 °C with a rabbit polyclonal anti-myeloperoxidase (MPO, a marker of neutrophils) antibody (1:200, Neomarkers, Fremont, CA, USA)[27], a mouse monoclonal anti-CD11b (a marker of macrophage/microglia) antibody (1:200, Serotec, Oxford, UK)[12], or a rabbit polyclonal antibody against 5-LOX (1:200, Cayman Chemical, Ann Arbor, MI, USA)[28] overnight at 4 °C, then incubated with biotinyl-ated goat anti-rabbit or goat anti-mouse IgG (1:200, Zhong-shan Biotechnology, Beijing, China) for 2 h at room tempera-ture, and horseradish peroxidase-streptavidin (1:200, Zhong-shan Biotechnology, China) for 60 min. Finally, the sections were exposed for 5–20 min to 0.05% 3, 3'-diamino-benzidine and 0.03% H2O2.
To visualize the localization of 5-LOX in different cell types, double immunofluorescence was employed. Briefly, after blocking non-specific binding of IgG with 5% normal goat serum for 2 h at room temperature, each section was incubated overnight at 4 °C with a mixture of a rabbit poly-clonal antibody against 5-LOX and a mouse monoclonal antibody against neuronal nuclear antigen (NeuN, 1:100, a specific marker of neurons, Chemicon, Temecula, CA, USA)[29], glial fibrillary acidic protein (GFAP, 1:800, a specific marker of astrocytes, Chemicon, USA)[29] or CD11b (1:200). Then, the sections were incubated with the mixture of fluorescein-isothiocyanate-conjugated goat anti-rabbit IgG and Cy3-conjugated goat anti-mouse IgG (Chemicon, USA). The anti-NeuN antibody alone was also used for detecting the neurons.
Endogenous IgG immunostaining was performed to detect blood-brain barrier (BBB) disruption[30]. Cortical sections (3.8–4.0 mm caudal to bregma) reacted respectively and successively with a biotinylated anti-rat IgG antibody (1:200, Sigma Chemical Co, St Louis, MO, USA), horseradish peroxidase-streptavidin (1:200) and 0.05% 3,3'-diamino-benzidine. The stained sections were photographed using a digital camera (FinePix S602 148 Zoom, Fuji, Tokyo, Japan), and analyzed with a computer using image analysis software (Imagetool 2.0, University of Texas Health Science Center, San Antonio, TX, USA). The percentage of immunoreactivity in the sections was determined as follows: [IgG-positive area in ischemic hemisphere/(contralateral hemisphere area)]×100%[30].
RT-PCR In another experiment, the cortical tissues were dissected on ice from the ischemic core and the boundary zone adjacent to the ischemic core 72 h after reperfusion, and stored at -70 °C until use. The control tissues were removed from the cortex of the sham-operated rats corresponding to the ischemic core. The total RNA was extracted using Trizol reagents (Invitrogen, Carlbad, CA, USA) according to the manufacture’s protocol. For the cDNA synthesis, 2 µg total RNA was mixed with 1 mmol/L dNTP, 0.2 µg random primer, 20 U RNasin, and 200 U M-MuLV reverse transcriptase in 20 µL reverse reaction buffer. The mixture was incubated at 42 ℃ for 60 min and then heated at 72 °C for 10 min to inactivate the reverse transcriptase.
PCR reactions were performed on an Eppendorf Master Cycler (Eppendorf, Hamburg, Germany). The reaction conditions were as follows: 1 µL cDNA mixture reacted in 20 µL reaction buffer containing 1.5 mmol/L MgCl2, 0.2 mmol/L dNTP, 20 pmol/L primer, and 1 U Taq DNA polymerase. The reactions were initially heated at 94 °C for 1 min, then at 94 °C for 30 s, 60 °C for 60 s, and 72 °C for 30 s, repeated for 35 cycles, and finally elongating at 72 °C for 10 min. Ten μL of the PCR products were separated by 2% agarose gel electrophoresis and visualized by ethidium bromide staining. The density of each band was measured by the UVP gel analysis system (Bio-Rad, Richimond, CA, USA). The mRNA expression of 5-LOX was reported as ratios of 5-LOX/β-actin. The primer sequences were as follows: 5-LOX, forward: 5'-AAA GAA CTG GAA ACA CGT CAG AAA-3', and reverse: 5'-AAC TGG TGT GTA CAG GGG TCA GTT-3'; β-actin, forward 5'-TCA TGA AGT GTG ACG TTG AC-3', and reverse 5'-CCT AGA AGC ATT TGC GGT GC-3'. The product sizes were 514 bp and 285 bp, respectively. The specificity of the primers was verified using the BLASTN program (http://www.ncbi.nlm.nih.gov/blast/).
Leukotriene measurement In a separate experiment, the ischemic cortexes were quickly isolated on ice 3 h after reperfusion, which was confirmed as the peak time of leukotriene production in our laboratory. The cortical tissues were weighed and homogenized in ice-cold absolute ethanol. After centrifugation of the homogenates at 15 000×g at 4 ℃ for 30 min, the supernatant was collected and filtrated through a 0.2 µm filtrator. The filtration was dried under nitrogen and resuspended in an ELISA buffer. The tissue contents of LTB4 and CysLT (the 5-LOX metabolites) were measured according to the protocol of the enzyme immunoassay kit (Cayman Chemical, USA). All measurements were carried out in duplicate.
Statistical analysis Data are expressed as the mean±SD. The differences between the groups were analyzed by one-way ANOVA followed by Student-Newman-Keuls t-test; the neurological deficit score and the percent area of IgG immunocreativity were analyzed by non-parametric Mann-Whitney U-test (SPSS 10.0 for Windows, 1999, SPSS, Chicago, IL, USA). A value of P<0.05 was considered statistically significant.
Results
Physiological parameters There were no significant differences in the mean arterial blood pressure, arterial blood PaO2, PaCO2, and blood glucose between 30 min before and 30 min after MCAO among the groups treated with saline and minocycline (22.5 and 45 mg/kg) as well as the sham-operation group. Treatment with minocycline did not affect these physiological parameters. In the ischemic rats, rCBF was reduced by approximately 50%–60% during 30 min MCAO and recovered to nearly baseline levels 15 min after reperfu-sion; treatment with minocycline did not alter rCBF reduction and recovery (Table 1).
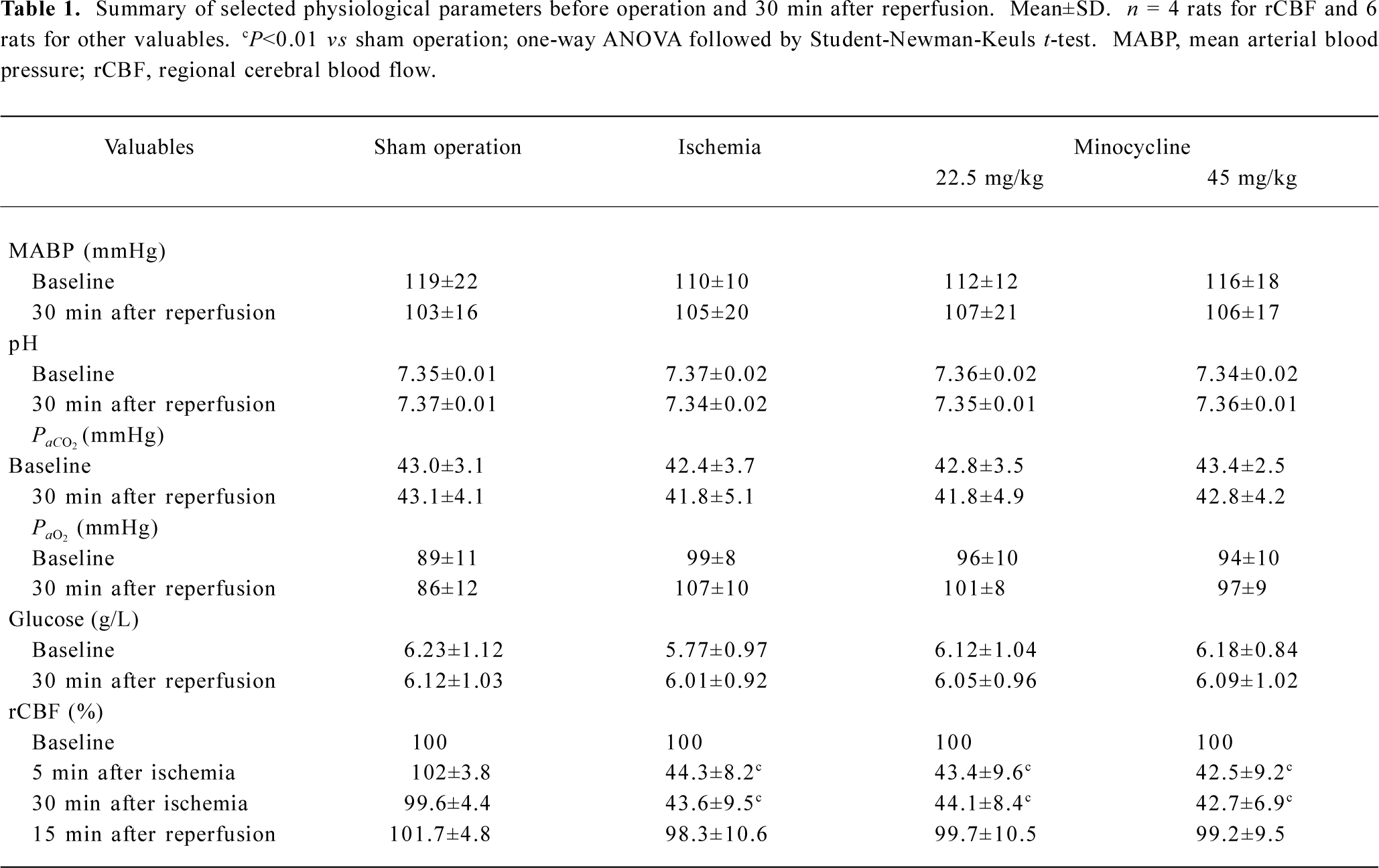
Full table
Ischemic injuries The neurological deficit score increased (Figure 1A) and the inclined degrees decreased significantly (Figure 1B) 72 h after reperfusion in the MCAO control rats. Minocycline (22.5 and 45 mg/kg) significantly reduced the neurological deficit scores (P<0.05 or 0.01 vs MCAO control, Figure 1A), and significantly increased the inclined degrees only at the concentration of 45 mg/kg (P<0.01 vs MCAO control, Figure 1B).
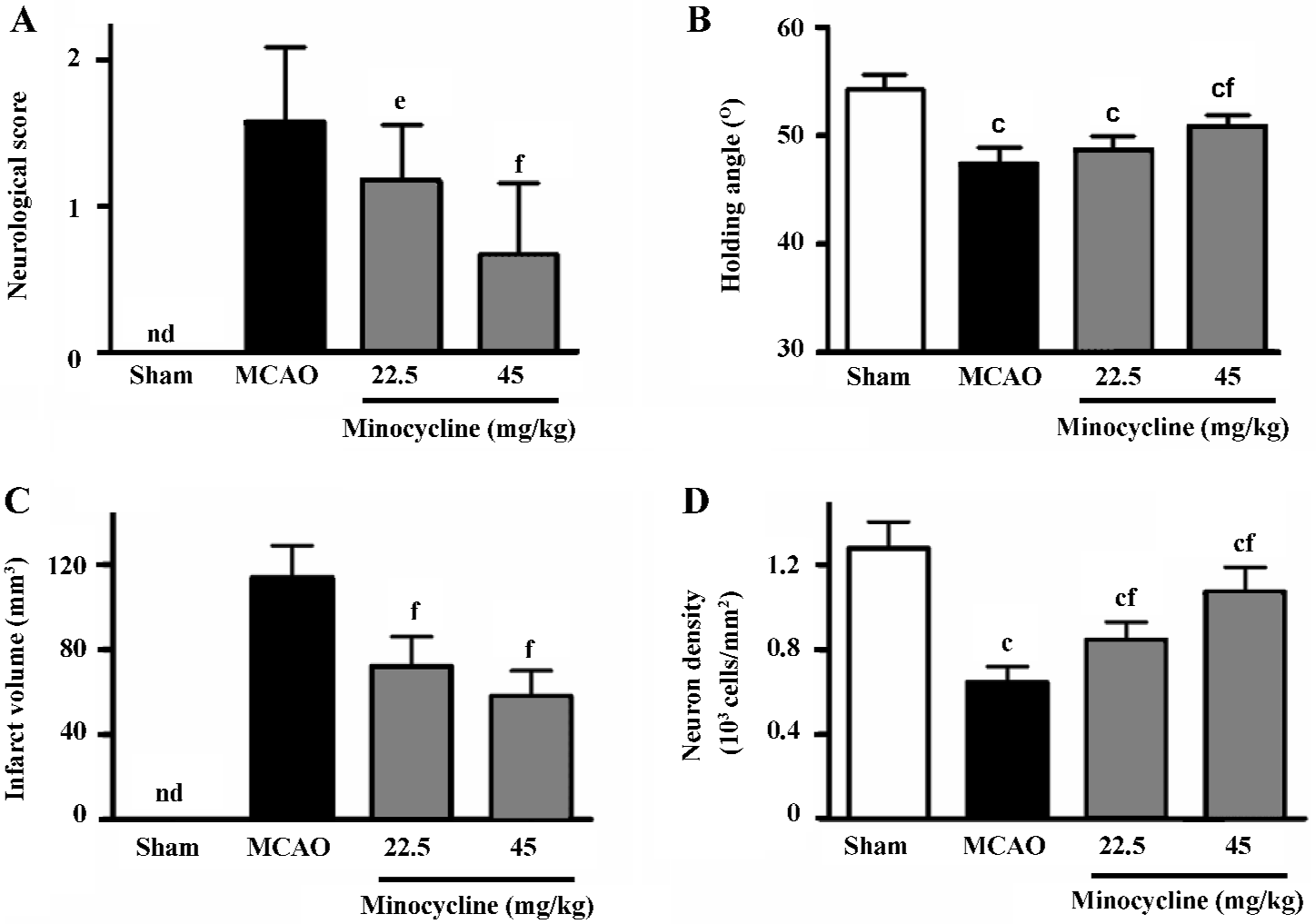
The infarct volume in the ischemic hemispheres was significantly reduced by minocycline (22.5 and 45 mg/kg) 72 h after reperfusion (P<0.01 vs MCAO control, Figure 1C). The reduced NeuN-positive neuron density in the boundary zone was significantly attenuated by minocycline (22.5 and 45 mg/kg) 72 h after reperfusion (P<0.01 vs MCAO control, Figure 1D).
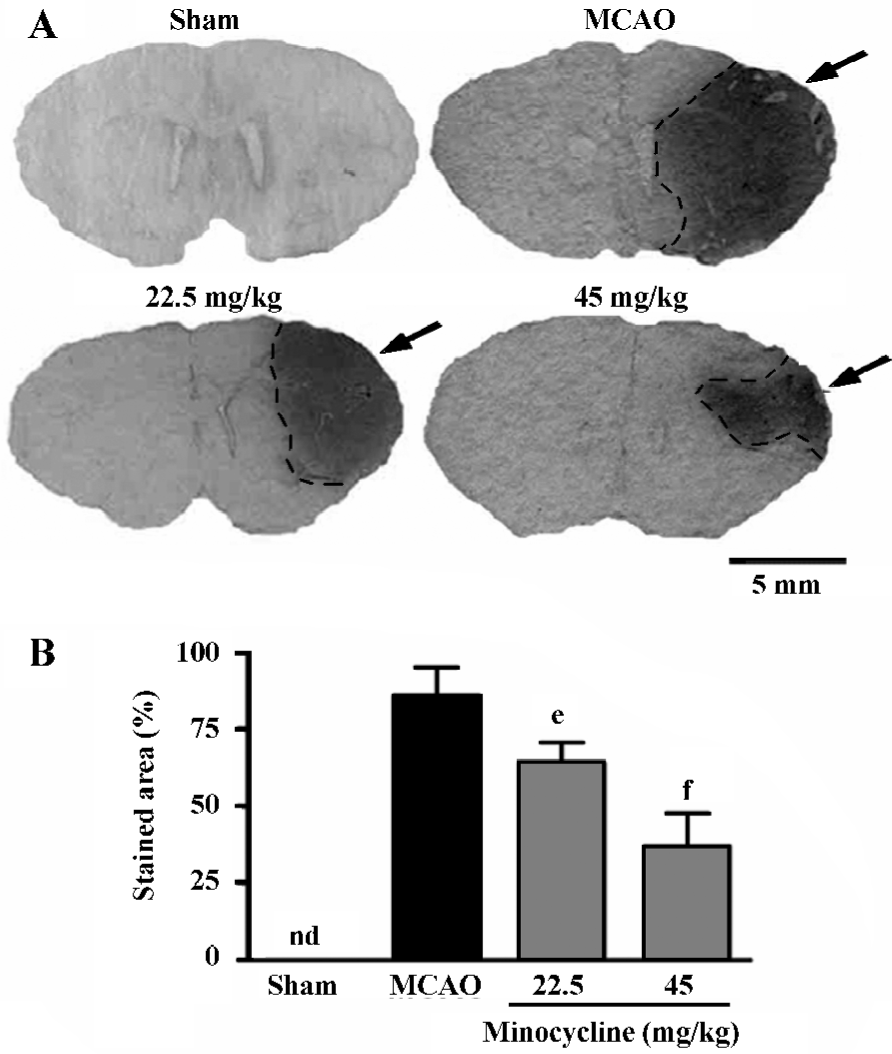
Inflammatory reactions The endogenous IgG immunoreactivity was intensively increased 72 h after reperfusion; minocycline (22.5 and 45 mg/kg) remarkably reduced IgG exudation (P<0.05 or 0.01 vs MCAO control, Figure 2).
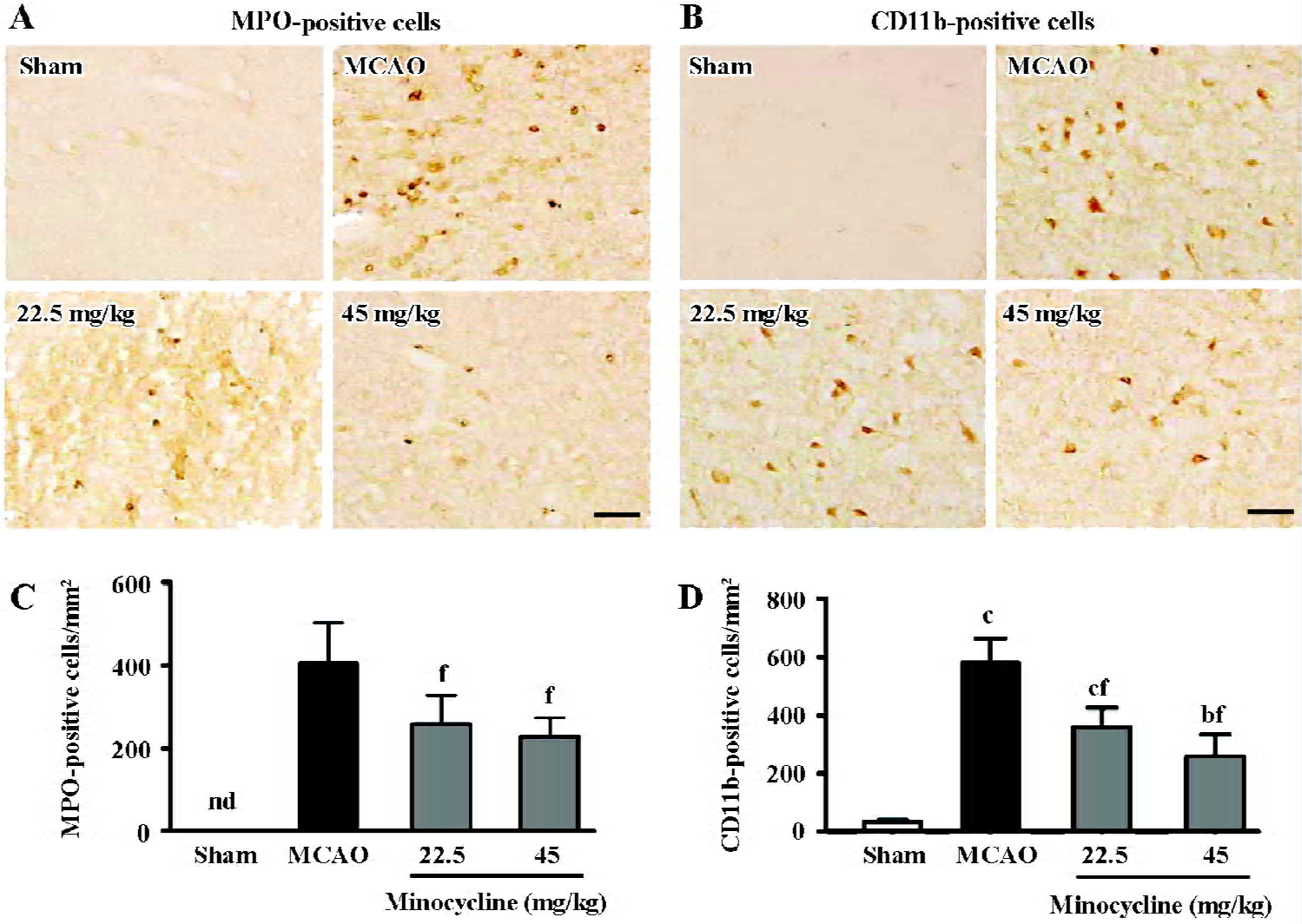
The amount of MPO-positive neutrophils and CD11b-positive macrophage/microglia increased in the ischemic hemispheres 72 h after reperfusion (Figure 3A, 3B). Mino-cycline (22.5 and 45 mg/kg) significantly inhibited neutrophil and macrophage/microglial accumulation (P<0.01 vs MCAO control, Figure 3C, 3D), as evidenced by MPO and CD11b immunoreactivity, respectively.
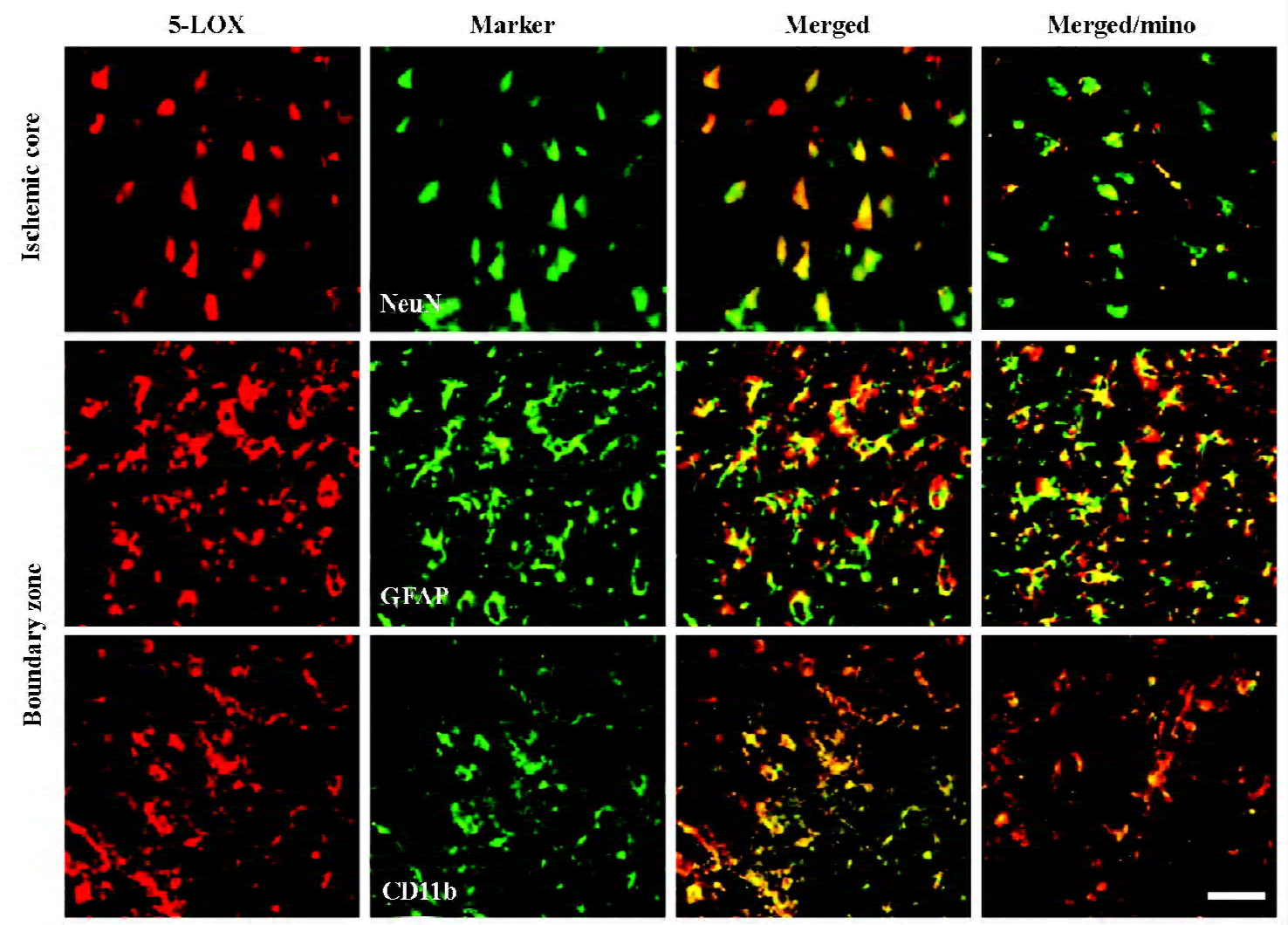
Changes in 5-LOX expression and enzymatic activity 5-LOX-positive cells were markedly increased in the ischemic core and boundary zone 72 h after reperfusion (Figure 4A), which was inhibited by minocycline (45 mg/kg, P<0.01, Figure 4B). Double immunofluorescence showed that 5-LOX was primarily localized in NeuN-positive neurons in the ischemic core, and in GFAP-positive astrocytes and CD11b-positive macrophage/microglia in the boundary zone (Figure 5); while 5-LOX expression was much weaker in astrocytes and macrophage/microglia in the ischemic core as well as in neurons in the boundary zone (data not shown). The 5-LOX-positive and CD11b-positive cells were reduced by mino-cycline (right panels in Figure 5).
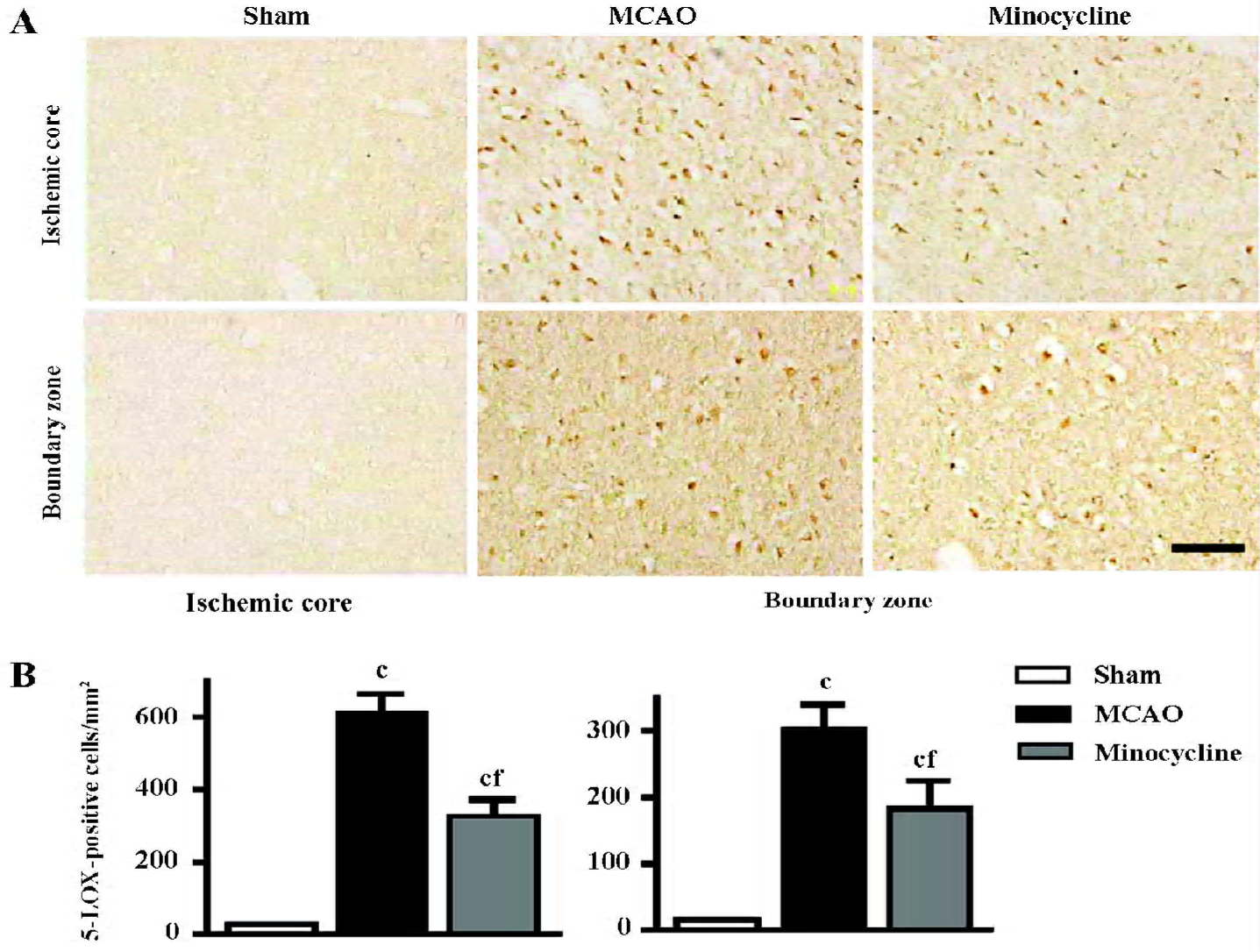
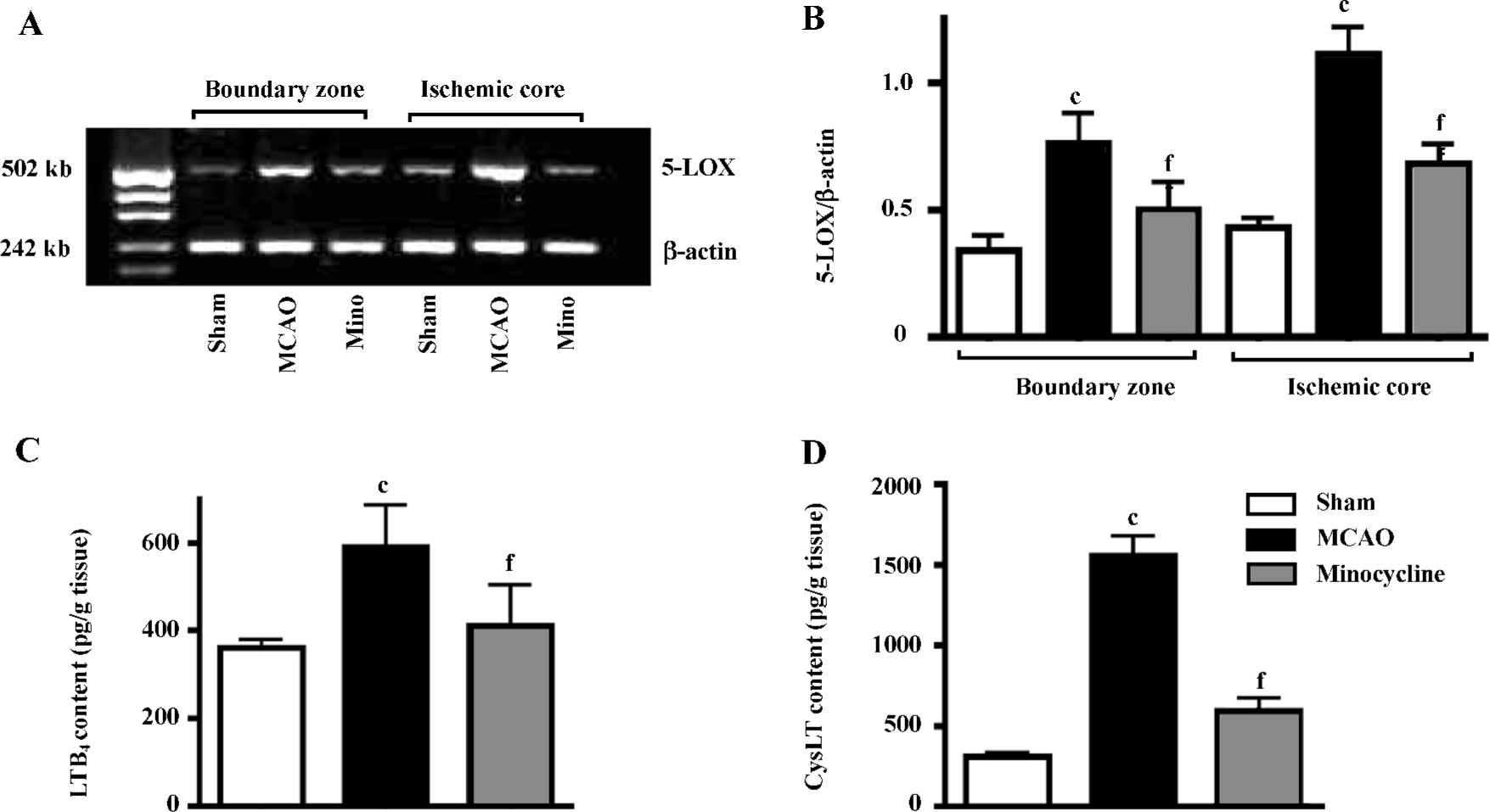
5-LOX mRNA expression was significantly increased in both the ischemic core and the boundary zone 72 h after reperfusion (Figure 6A), which was significantly inhibited by minocycline (45 mg/kg, P<0.01 vs MCAO control, Figure 6B). The contents of LTB4 and CysLT in the ischemic cortex increased 3 h after reperfusion, which was significantly reduced by minocycline (45 mg/kg, P<0.01 vs MCAO control, Figure 6C, 6D).
Discussion
In the present study, we found that minocycline exerted the anti-inflammatory effects after ischemia/reperfusion injury in rats as previously reported[5–10]. Minocycline inhibits 5-LOX expression and activation in ischemic brain tissue after focal cerebral ischemia, confirming its ability in PC12 cells[19,20]. These findings indicate that minocycline might inhibit postischemic inflammation via modulating 5-LOX expression and activation.
In postischemic brain inflammation in the subacute phase (days after ischemia), the brain-blood barrier (BBB) disruption, as well as the accumulation of neutrophils and macrophage/microglia in the brain, are the determinants in pathogenesis[31,32]. The present study indicates that minocycline inhibits endogenous IgG exudation, an indicator of BBB disruption, as previously reported[30]. This effect of minocycline might result from the inhibition of neutrophil accumulation because the disrupted BBB has been reported to promote blood leukocyte recruitment[33]. However, resident microglia and hematogenous macrophages play similar roles in the pathogenetic cascade after cerebral ischemia; both are CD11b-positive, but the distinction between these cells has not been possible due to a lack of discriminating cellular markers[34]. Since minocycline inhibits microglial activation[11,12] and BBB disruption, it might directly inhibit resident microglia activation and/or indirectly result from the inhibition of BBB disruption.
The most important finding in the present study is that minocycline may inhibit postischemic inflammation by inhibiting 5-LOX expression and enzymatic activation. Similarly, we also found that minocycline inhibited 5-LOX activation and exerted a protective effect on in vitro ischemic-like and excitotoxic injury in PC12 cells[19,20]. 5-LOX activation has been reported in transient global ischemia[5,6] and focal cerebral ischemia[7,9], and in the human brain tissue from stroke patients[8]. Recently, we found that 5-LOX expression and enzymatic activity increased after focal cerebral ischemia in rats, and were spatio-temporally involved in neuron injury in the acute phase and astrocyte proliferation in the late phase in vivo[10]. These findings indicate that 5-LOX may be a target of minocycline neuroprotection. Mino-cycline inhibits the activation of p38 mitogen-activated protein kinase (MAPK)[35,36], which may explain its effect on 5-LOX translocation or activation. As shown in neutrophils, activated p38 MAPK phosphorylates MAPK-activated protein kinase 2 and 3, which then directly phosphorylates 5-LOX; the phosphorylated 5-LOX translocates to the nuclear membrane and is activated to produce leukotrienes[37]. There-fore, minocycline possibly inhibits 5-LOX activation by modulating p38 MAPK activity. However, we could not explain why minocycline reduces the expression of 5-LOX, which may result from secondary responses due to attenuation of inflammation or from a special ability of minocycline.
The inhibition of 5-LOX activation may explain the anti-inflammatory effects of minocycline. As supporting evidence, microglia, neutrophils, and macrophages possess the leukotriene-producing capacity of 5-LOX[38–40]. The 5-LOX metabolites, leukotrienes (LTB4 and CysLT), are potent inflammatory mediators that can induce a variety of res-ponses, such as chemotaxis of leukocytes[41] and BBB disruption[25,42]. Therefore, reduction of leukotriene production by minocycline might attenuate postischemic inflammatory reactions. Additionally, minocycline possesses many other abilities that also relate to its anti-inflammatory effects, such as the inhibition of cytokine production[43], iNOS[43], metallo-protease activity[13], and antioxidant ability[14].
However, as discordant evidence, no difference in ischemic infarcts has been found between 5-LOX-deficient and wild-type mice with focal cerebral ischemia[44]. We explained the discrepancy by 2 possibilities: one possibi-lity is that 5-LOX deficiency may cause compensations as reported otherwise[45,46], which should be considered as an explanation for the results from wild-type and 5-LOX-defici-ent animals. Another possibility is that 5-LOX may act only as one factor in a complex system on ischemic brain injury; even if in arachidonic acid-metabolizing pathway, 5-LOX deficiency may enhance cyclooxygenase activity[47]. Recent-ly, 5-LOX was confirmed as a modulator in rat focal cerebral ischemia in another laboratory[48]. Therefore, 5-LOX activation may be reasonably considered as one of the targets of minocycline.
In conclusion, we found that minocycline inhibited postischemic inflammation, partly mediated by the inhibition of 5-LOX expression and enzymatic activation.
References
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab 1999;19:819-34.
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999;22:391-7.
- Helgadottir A, Manolescu A, Thorleifsson G, Gretarsdottir S, Jonsdottir H, Thorsteinsdottir U, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet 2004;36:233-9.
- Lohmussaar E, Gschwendtner A, Mueller JC, Org T, Wichmann E, Hamann G, et al. ALOX5AP gene and the PDE4D gene in a central European population of stroke patients. Stroke 2005;36:731-6.
- Ohtsuki T, Matsumoto M, Hayashi Y, Yamamoto K, Kitagawa K, Ogawa S, et al. Reperfusion induces 5-lipoxygenase translocation and leukotriene C4 production in ischemic brain. Am J Physiol 1995;268:H1249-57.
- Baskaya MK, Hu Y, Donaldson D, Maley M, Rao AM, Prasad MR, et al. Protective effect of the 5-lipoxygenase inhibitor AA-861 on cerebral edema after transient ischemia. J Neurosurg 1996;85:112-6.
- Ciceri P, Rabuffetti M, Monopoli A, Nicosia S. Production of leukotrienes in a model of focal cerebral ischaemia in the rat. Br J Pharmacol 2001;133:1323-9.
- Tomimoto H, Shibata M, Ihara M, Akiguchi I, Ohtani R, Budka H. A comparative study on the expression of cyclooxygenase and 5-lipoxygenase during cerebral ischemia in humans. Acta Neuropathol (Berl) 2002;104:601-7.
- Zhang RL, Lu CZ, Ren HM, Xiao BG. Metabolic changes of arachidonic acid after cerebral ischemia-reperfusion in diabetic rats. Exp Neurol 2003;184:746-52.
- Zhou Y, Wei EQ, Fang SH, Chu LS, Wang ML, Zhang WP, et al. Spatio-temporal properties of 5-lipoxygenase expression and activation in the brain after focal cerebral ischemia in rats. Life Sci 2006;79:1645-56.
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotec-tive in global brain ischemia. Proc Natl Acad Sci USA 1998;95:15769-74.
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA 1999;96:13496-500.
- Koistinaho M, Malm TM, Kettunen MI, Goldsteins G, Starckx S, Kauppinen RA, et al. Minocycline protects against permanent cerebral ischemia in wild type but not in matrix metalloprotease-9-deficient mice. J Cereb Blood Flow Metab 2005;25:460-7.
- Morimoto N, Shimazawa M, Yamashima T, Nagai H, Hara H. Minocycline inhibits oxidative stress and decreases in vitro and in vivo ischemic neuronal damage. Brain Res 2005;1044:8-15.
- Hewlett KA, Corbett D. Delayed minocycline treatment reduces long-term functional deficits and histological injury in a rodent model of focal ischemia. Neuroscience 2006;141:27-33.
- Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery 2001;48:1393-9.
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002;417:74-8.
- Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, . Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc Natl Acad Sci USA 2003; 100: 10 483–7.
- Song Y, Wei EQ, Zhang WP, Zhang L, Liu JR, Chen Z. Mino-cycline protects PC12 cells from ischemic-like injury and inhibits 5-lipoxygenase activation. Neuroreport 2004;15:2181-4.
- Song Y, Wei EQ, Zhang WP, Ge QF, Liu JR, Wang ML, et al. Minocycline protects PC12 cells against NMDA-induced injury via inhibiting 5-lipoxygenase activation. Brain Res 2006;1085:57-67.
- Yano T, Anraku S, Nakayama R, Ushijima K. Neuroprotective effect of urinary trypsin inhibitor against focal cerebral ischemia-reperfusion injury in rats. Anesthesiology 2003;98:465-73.
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989;20:84-91.
- Prestigiacomo CJ, Kim SC, Connolly ES Jr, Liao H, Yan SF, Pinsky DJ. CD18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke 1999;30:1110-7.
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke 1986;17:472-6.
- Yu GL, Wei EQ, Zhang SH, Xu HM, Chu LS, Zhang WP, et al. Montelukast, a cysteinyl leukotriene receptor-1 antagonist, dose- and time-dependently protects against focal cerebral ischemia in mice. Pharmacology 2005;73:31-40.
- Yonemori F, Yamaguchi T, Yamada H, Tamura A. Evaluation of a motor deficit after chronic focal cerebral ischemia in rats. J Cereb Blood Flow Metab 1998;18:1099-106.
- Hua R, Walz W. Minocycline treatment prevents cavitation in rats after a cortical devascularizing lesion. Brain Res 2006;1090:172-81.
- Zuo L, Christofi FL, Wright VP, Bao S, Clanton TL. Lipoxygen-ase-dependent superoxide release in skeletal muscle. J Appl Physiol 2004;97:661-8.
- Schmidt W, Reymann KG. Proliferating cells differentiate into neurons in the hippocampal CA1 region of gerbils after global cerebral ischemia. Neurosci Lett 2002;334:153-6.
- Muramatsu K, Fukuda A, Togari H, Wada Y, Nishino H. Vulnerability to cerebral hypoxic-ischemic insult in neonatal but not in adult rats is in parallel with disruption of the blood–brain barrier. Stroke 1997;28:2281-8.
- Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol 2003;62:127-36.
- Simundic AM, Basic V, Topic E, Demarin V, Vrkic N, Kunovic B, et al. Soluble adhesion molecules in acute ischemic stroke. Clin Invest Med 2004;27:86-92.
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, et al. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol 2005;289:H558-68.
- Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: A study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 2003;183:25-33.
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol 2001;166:7527-33.
- Lin S, Zhang Y, Dodel R, Farlow MR, Paul SM, Du Y. Minocycline blocks nitric oxide-induced neurotoxicity by inhibition p38 MAP kinase in rat cerebellar granule neurons. Neurosci Lett 2001;315:61-4.
- Werz O, Klemm J, Samuelsson B, Radmark O. 5-Lipoxygenase is phosphorylated by p38 kinase-dependent MAPKAP kinases. Proc Natl Acad Sci USA 2000;97:5261-6.
- Brock TG, McNish RW, Peters-Golden M. Translocation and leukotriene synthetic capacity of nuclear 5-lipoxygenase in rat basophilic leukemia cells and alveolar macrophages. J Biol Chem 1995; 270: 21 652–8.
- Brock TG, McNish RW, Bailie MB, Peters–Golden M. Rapid import of cytosolic 5-lipoxygenase into the nucleus of neutrophils after in vivo recruitment and in vitro adherence. J Biol Chem 1997;272:8276-80.
- Ballerini P, Di Iorio P, Ciccarelli R, Caciagli F, Poli A, Beraudi A, et al. P2Y1 and cysteinyl leukotriene receptors mediate purine and cysteinyl leukotriene co-release in primary cultures of rat microglia. Int J Immunopathol Pharmacol 2005;18:255-68.
- Barone FC, Hillegass LM, Tzimas MN, Schmidt DB, Foley JJ, White RF, et al. Time-related changes in myeloperoxidase activity and leukotriene B4 receptor binding reflect leukocyte influx in cerebral focal stroke. Mol Chem Neuropathol 1995;24:13-30.
- Baba T, Black KL, Ikezaki K, Chen KN, Becker DP. Intracarotid infusion of leukotriene C4 selectively increases blood-brain barrier permeability after focal ischemia in rats. J Cereb Blood Flow Metab 1991;11:638-43.
- Lai AY, Todd KG. Hypoxia-activated microglial mediators of neuronal survival are differentially regulated by tetracyclines. Glia 2006;53:809-16.
- Kitagawa K, Matsumoto M, Hori M. Cerebral ischemia in 5-lipoxygenase knockout mice. Brain Res 2004;1004:198-202.
- Takahashi E, Ino M, Miyamoto N, Nagasu T. Increased expression of P/Q-type Ca2+ channel alpha1A subunit mRNA in cerebellum of N-type Ca2+ channel alpha1B subunit gene-deficient mice. Brain Res Mol Brain Res 2004;124:79-87.
- Snell BJ, Day A, Ledent C, Lawrence AJ. [3H]Adenosine uptake in brainstem membranes of CD-1 mice lacking the adenosine A(2a) receptor. Life Sci 2004;75:225-35.
- Goulet JL, Snouwaert JN, Latour AM, Coffman TM, Koller BH. Altered inflammatory responses in leukotriene-deficient mice. Proc Natl Acad Sci USA 1994;91:12852-6.
- Jatana M, Giri S, Ansari MA, Elango C, Singh AK, Singh I, et al. Inhibition of NF-kappaB activation by 5-lipoxygenase inhibitors protects brain against injury in a rat model of focal cerebral ischemia. J Neuroinflammation 2006;3:12.