
Requirement of PSD-95 for dopamine D1 receptor modulating glutamate NR1a/NR2B receptor function1
IntroductionOther Section
The dopamine (DA) D1 receptor (D1R) and the glutamate (Glu) NMDA receptor (NMDAR) represent two functionally and structurally diverse receptor classes. D1R belongs to G-protein-coupled receptor family and couples to Gαs proteins. NMDAR are ligand-gated ion channels composed of multiple subunits (NR1, NR2A-D, and NR3A-C)[1]. A remarkable property of NMDAR is its high permeability to Ca2+. Alteration of NMDAR-mediated Ca2+ influx is reported to be involved in schizophrenia and in excitotoxic neuronal death associated with brain disorders such as stroke, epilepsy, and trauma[2].
D1R and NMDAR are co-localized in several brain structures, including caudate-putamen, nucleus accumbens, hippocampus, and prefrontal cortex[3–5]. D1R and NMDAR are constitutively interacting in cells as revealed by co-immunoprecipitation and fluorescence resonance energy transfer (FRET) studies[6]. The direct physical interaction is mediated through the C terminal of the respective protein[7]. In addition to the direct interaction at the receptor level, previous studies have also shown that the downstream signal molecules mediated by D1R is also found to regulate NMDAR function. For instance, D1R stimulated protein kinase A (PKA) was demonstrated to phosphorylate the NR1 subunit of NMDAR[8] and to enhance NMDA-mediated excitability[9]. Furthermore, the activation of D1R was also found to enhance NMDA currents via protein kinase C (PKC)-dependent mechanisms[10]. However, the direct D1R-NMDAR interaction was also reported to result in the inhibition of NMDAR-mediated currents[8], indicating that the D1-NMDAR interaction is rather a diverse and complex event.
D1R and NMDAR are presented at high concentrations in the postsynaptic density (PSD)[6]. PSD is a highly organized subcellular fraction in which NMDAR co-exists with scaffolding proteins such as PSD-95 and other signaling proteins[11]. PSD-95 comprises three primary decidual zone (PDZ) domains, a Src-homology three domain, and a domain homologous to guanylate kinase[12]. It is known that PSD-95 utilizes those domains for protein interaction to recruit signaling proteins and to mediate its assembly with other components of the PSD.
Recent information indicates that PSD-95 can regulate membrane trafficking and intracellular signaling of a number of neurotransmitter receptors including NMDA, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA), and 5-hydroxytryptamine 2A (5-HT2A) receptors via physical interactions[13–15]. As D1R and NMDAR are presented at high density in the PSD[6], it is conceivable that PSD-95 may also regulate D1/NMDA receptor interaction. To elucidate the role of PSD-95 in D1R-modulated functions of the NR1a/NR2B receptor, HEK293 cells expressing D1R and NR1a/NR2B receptors were employed to study the role of PSD-95 in the D1R-modulated NR1a/NR2B receptor-mediated Ca2+ influx. Our study demonstrated that PSD-95 was required in D1R modulating NR1a/NR2B receptor function, and this modulatory effect depended on PKA and PKC.
Materials and methodsOther Section
Materials Fura-2 acetoxymethyl ester (Fura-2 AM) was from Molecular Probe (Eugene, OR, USA). 3-Isobutyl-1-methyl-2,6 (1H, 3H)-purinedione (IBMX) and Pluronic-127, monoclonal anti-D1 DA receptor antibody produced in rat clone 1-1-F11 s.E6, monoclonal anticmyc antibody produced in mouse clone 9E10, anti-mouse IgG (whole molecule)-TRITC antibody produced in goats, anti-HA antibody produced in rabbits, leupeptin, pepstatin A, aprotinin, and phenyl-methanesulfonyl fluoride (PMSF) were purchased from Sigma (St Louis, MO, USA). Normal mouse IgG-HRP (horseradish peroxidase) was from Santa Cruz (Santa Cruz, CA, USA), anti-mouse IgG (H+L)-AP was from Promega (Madison, WI, USA), and [3H]SCH23390 was from Amersham (Cleveland, OH, USA). l-Glutamate acid sodium salt and glycine (Gly) were from Sino-American Biotech (Beijing, China). (±)-SKF-38393 hydrochloride, DA, H89, 8-Br-cAMP, and chelerythrine were from RBI (Natick, MA, USA). The cAMP assay kit was from the Shanghai University of Traditional Chinese Medicine (Shanghai, China). Other reagents were obtained as indicated in the text.
Cell culture and transfection Human embryonic kidney 293 (HEK293) cells, a generous gift from Dr Gang PEI (Institute of Biochemistry and Cell Biology, Chinese Academy of Science, Shanghai, China), were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% newborn calf serum (SiJiQing, Hangzhou, China), penicillin (100 U/mL), and streptomycin (100 U/mL). 2.5×104 cells were plated on poly-L-lysine-coated glass coverslips. Transfections were performed while cells reached 80% confluence. NR1a, NR2B, D1-enhanced yellow fluorescent protein (EYFP), and PSD-95 were delivered at a ratio of 1:1:1:1 by the calcium-phosphate transfection method. Ketamine 1 mmol/L was added to the culture dish to prevent excitotoxicity during transfection. If not indicated specifically, the cells used for the experiments were harvested 24 h after transfection. NR1a and NR2B cDNA were generous gifts from Dr John WOODWARD (Medical University of South Carolina, Charleston, USA), cDNA for PSD-95 were from Dr Morgan SHENG(Harvard University, Boston, MA, USA), and D1-EYFP was constructed by Dr You HE in our Laboratory.
Assays of cAMP content After 18–24 h transfection with plasmid-encoded D1-EYFP receptors, the cells were reseeded into a 96-well plate (1×104 cells/well) for 12 h. The cells were pre-incubated with 50 µL serum free DMEM containing 500 µmol/L IBMX prior to D1R agonist R-(+)-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine-7,8-diol hydrochloride (SKF38393) stimulation for an additional 10 min. The reaction was then terminated on ice by adding 100 µL 1 mol/L trichloroacetic acid. Following the addition of 20 µL 2 mol/L K2CO3, the sample was centrifuged for 5 min at 12 000×g. The supernatant was kept (diluted in 1:10) for determining cAMP content. All experiments were performed in duplicate, and each experiment was repeated at least 3 times.
Internalization assays Cells expressing D1-EYFP were treated either with 10 µmol/L DA or vehicle for 30 min in serum-free DMEM and then washed 3 times with ice-cold phosphate-buffered saline (PBS). The cells were fixed in 4% paraformaldehyde for 20 min at room temperature before 3 washes of PBS. The receptor internalization was then observed with a Leica SP2 confocal microscope (Leica microsystem, Heideberg, Germany). The excitation wavelength for yellow fluorescent protein (YFP) is at 514 nm.
Calcium imaging Transfected cells were incubated with Fura-2 AM (4 µmol/L with 0.025% Pluronic-127) for 45 min in extracellular solution (135 mmol/L NaCl, 5.4 mmol/L KCl, 1.8 mmol/L CaCl2, 10 mmol/L glucose, and 5 mmol/L HEPES (2-[4-(2-Hydroxyethyl)-1-piperazinyl]ethanesulfonic acid), pH 6.8). After 3 washes with Fura-free extracellular solution (pH 7.2), the dishes were mounted onto the stage of an Olympus BX51WI upright microscope (Tokyo, Japan). The cells were perfused continuously at a flow rate of 1.2–1.5 mL/min. Glu/Gly were applied via a computer-controlled Y-tube (outer φ=100 µm) situated 80 µm above the cells. The cells were exposed to alternating 340 nm and 380 nm light every 2 s during and immediately after Glu/Gly application. The signals were acquired via a charge coupled device (CCD) camera (CoolSNAP HQ, Roper Scientific, Duluth, GA, USA). To minimize UV exposure, images were taken every 15–60 s between drug applications. Ratio images were generated with MetaFlour software from Universal Imaging (West Chester, PA, USA). Intracellular calcium concentrations were determined by the ratio of the intensity of 340/380. NMDAR-dependent increases in intracellular calcium were calculated by subtracting the average baseline value from the peak value obtained during Glu/Gly application.
Data analysis All data were expressed as mean±SEM. Unless otherwise indicated, the statistical significance was determined using the least significant difference (LSD) test following ANCOVA with SPSS 11.0 (SPSS, Chicago, USA). The covariate was the average baseline NMDA response before perfusion of the antipsychotic drugs.
ResultsOther Section
Establishing and characterizing D1R-NR1a/NR2B receptor co-expression in HEK293 cells The HEK293 cells were co-transfected with D1-EYFP and the NR1a/NR2B receptor. To verify the success of transfection, the cells were locally perfused with Glu/Gly (100/10 µmol/L) for 3 s. This treatment induced reproducible Ca2+ influx (Figure 1A, upper panel). The application of 1 µmol/L MK-801 significantly blocked Glu/Gly-induced Ca2+ influx (Figure 1A, lower panel). Employing confocal microscopy, we observed the D1-EYFP receptor location on the cell membrane (Figure 1B, upper left). The application of 10 µmol/L DA to the cells induced significant internalization of D1R (Figure 1B, upper right). Moreover, SKF38393 resulted in an increase in cAMP accumulation in a dose-dependent manner (Figure 1B, lower panel). The mean EC50 value of SKF38393 was 0.16 µmol/L, similar to a previous report[16]. Thus, D1R and the NR1a/NR2B receptor were functionally expressed in HEK293 cells.
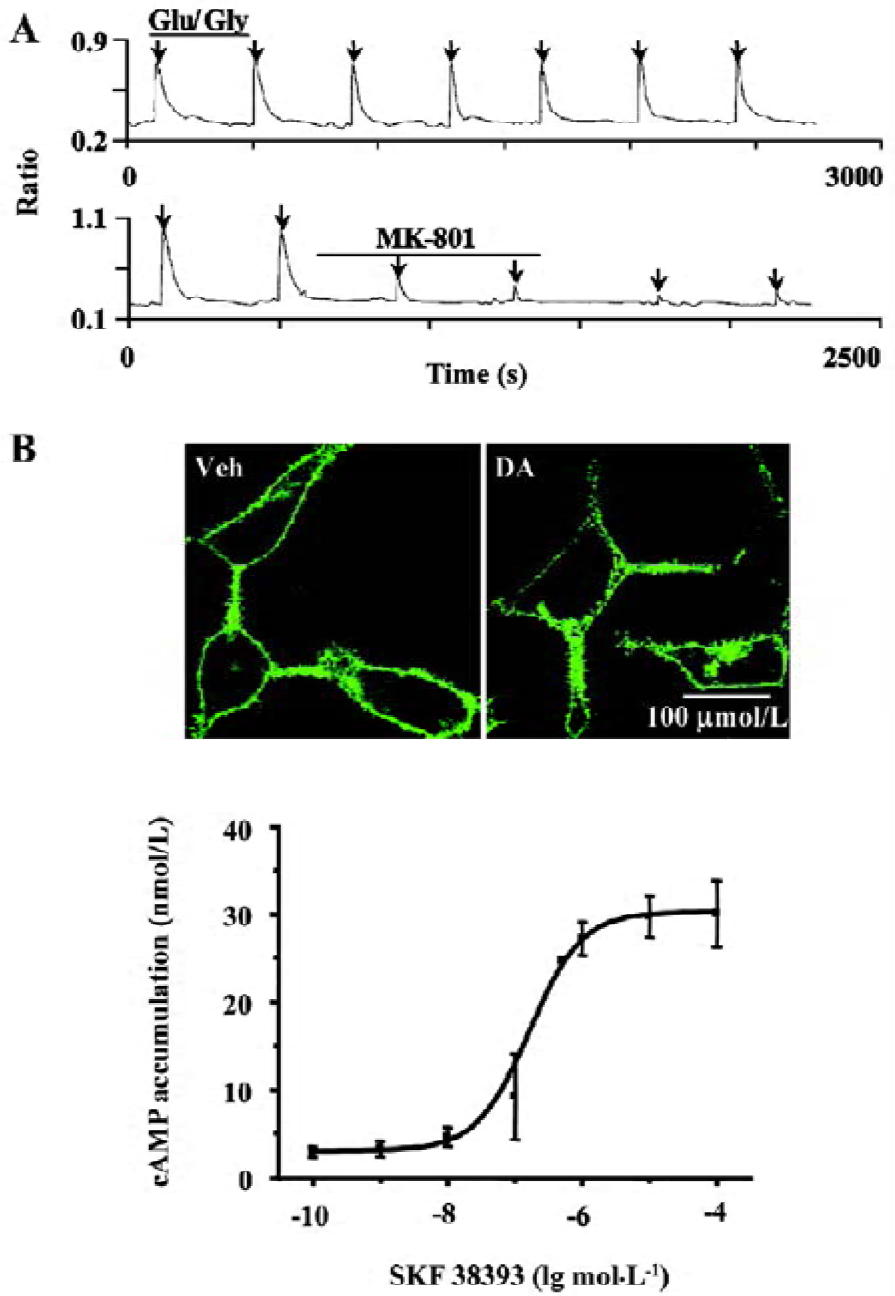
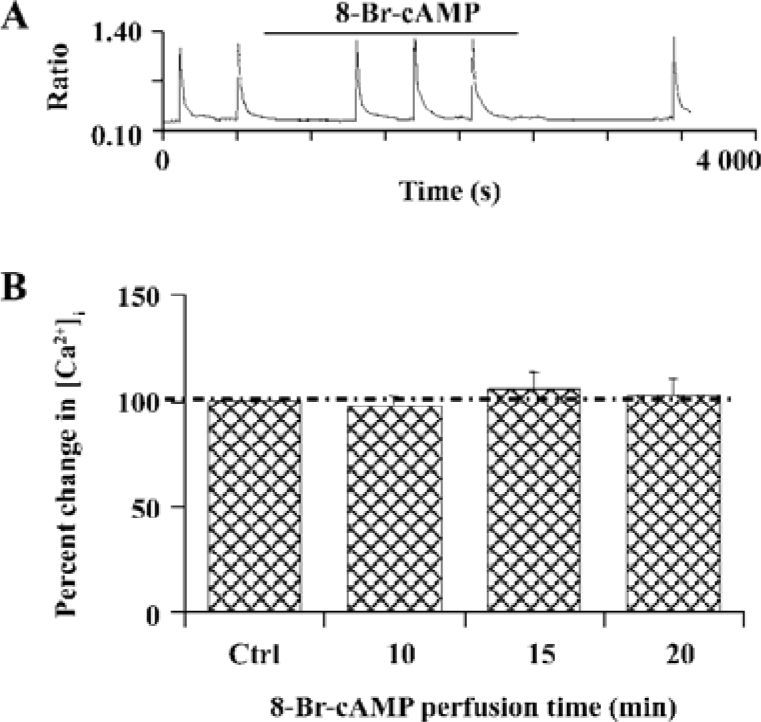
D1R activation did not affect NR1a/NR2B-mediated Ca2+ influx in HEK293 cells that expressed D1R and NR1a/NR2B We next tested whether D1R activation could modulate NR1a/NR2B receptor-mediated Ca2+ influx in HEK293 cells expressing both the D1R and NR1a/NR2B receptor. Glu/Gly-induced Ca2+ influx in the presence of DA (100 µmol/L) is shown in Figure 2A, 2B. The results clearly indicated that the activation of D1-EYFP did not alter NR1a/NR2B-mediated Ca2+ influx in our system. To exclude the possibility that the EYFP tag interrupts the interaction between the D1R and the NR1a/NR2B receptor, non-tagged D1R-NR1a/NR2B receptor co-transfected cells were tested. Again, the activation of D1R did not alter NR1a/NR2B-mediated Ca2+ influx as well in the cells (Figure 2A, 2B, right). Thus, it is clear that D1R is unable to modulate NR1a/NR2B receptor function in our in vitro co-expression system. In order to test whether the lack of D1R modulation on NR1a/NR2B-mediated response as described earlier was due to insufficient PKA activation by D1R in the system, the cell permanent PKA activator 8-Br-cAMP was used. Incubation of the cells with 10 µmol/L 8-Br-cAMP elicited no effect on the Glu/Gly-induced Ca2+ influx in HEK293 cells expressing both the D1R and the NR1a/NR2B receptor (Figure 3). Taken together, our data indicate that the activation of D1R or PKA fails to modulate NR1a/NR2B receptor-mediated Ca2+ influx in HEK293 cells expressed with the D1R and the NR1a/NR2B receptor.
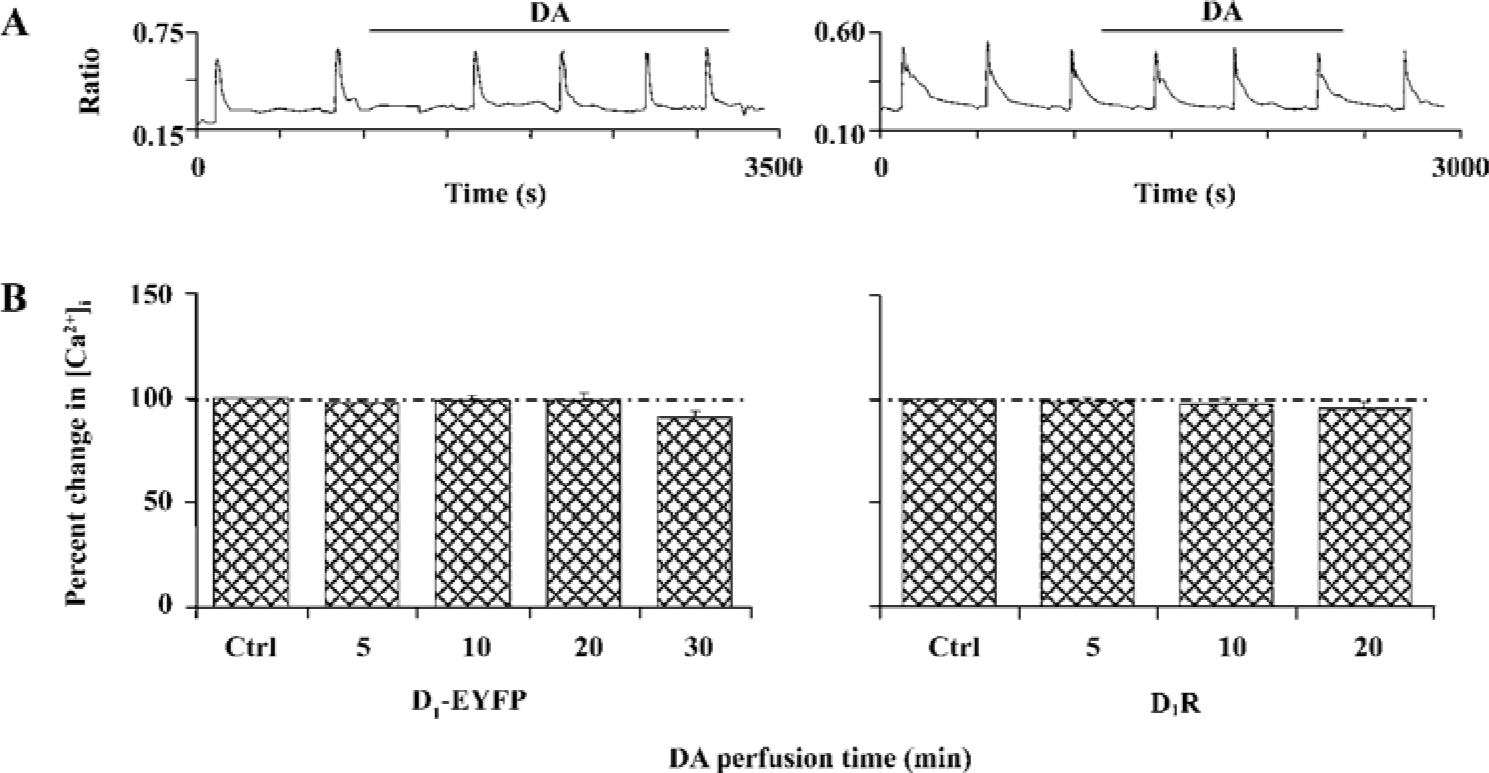
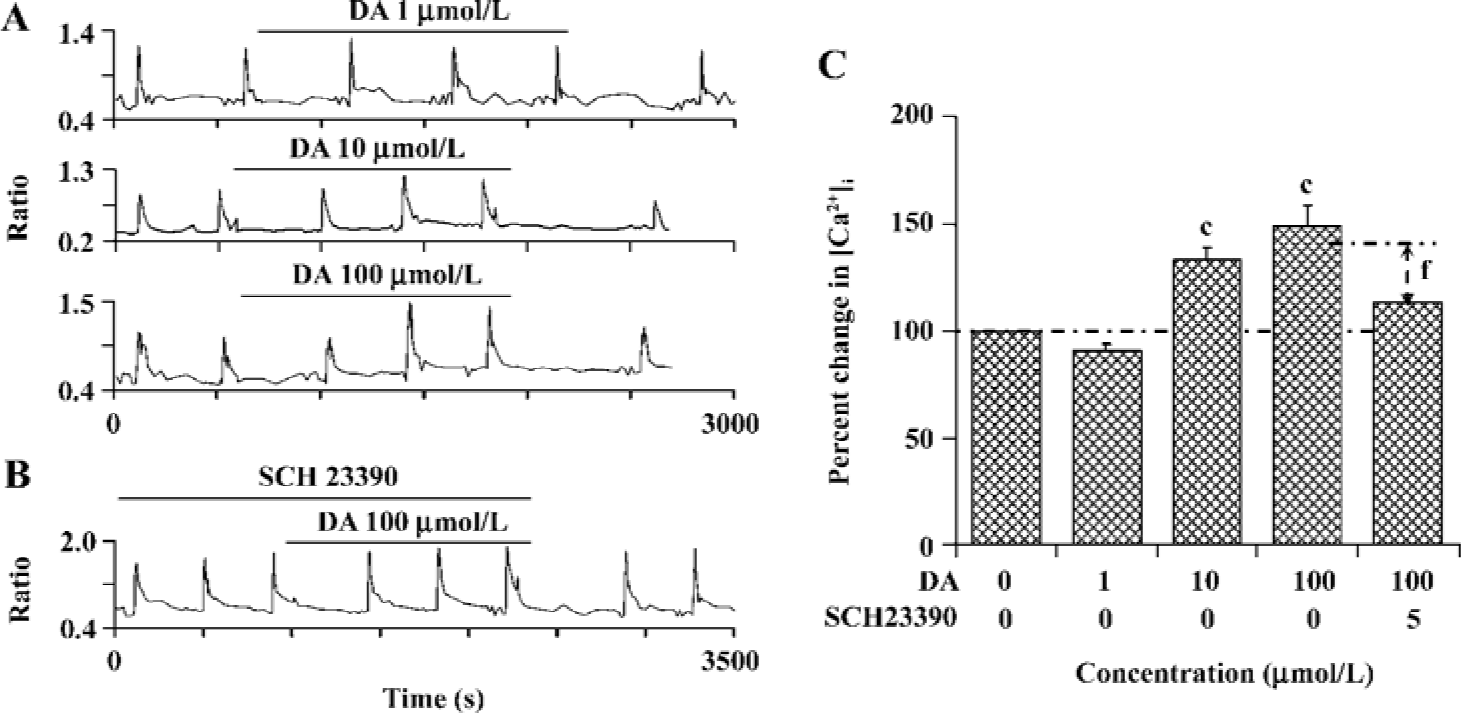
PSD-95 is required for D1R modulation of NR1a/NR2B receptor function Since it has been shown that the scaffold protein PSD-95 is associated with NMDAR and is involved in the regulation of receptor function, we wondered whether D1R activation could modulate NR1a/NR2B receptor function when PSD-95 is presented in our expression system. Interestingly, after co-expression with PSD-95, activation of D1R by DA led to an enhancement in NR1a/NR2B receptor function in a concentration-dependent manner. DA 10 µmol/L or 100 µmol/L induced a 34.0%±5.3% (F1,37=64.95, n=20, P<0.01) or a 48.6%±10.3% (F1,97=67.58, n=50, P<0.01) increase in Ca2+ influx, respectively (Figure 4A). As expected, the enhanced NR1a/NR2B receptor function induced by DA was blocked by 5 µmol/L SCH23390, a selective D1R antagonist (DA vs DA+SCH23390, F1,65=3.71, n=67, P<0.01, independent-samples t-test, Figure 4B,C), indicating a D1R-mediated event. This result demonstrated clearly that PSD-95 was required for the modulatory effect of D1R on NR1a/NR2B receptor function.
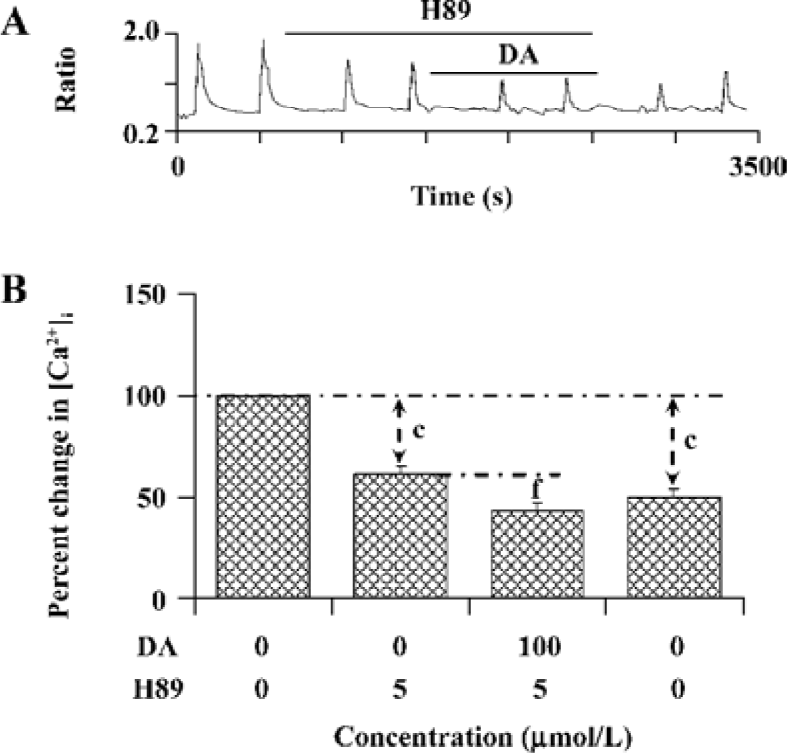
PKA is involved in the modulation of NR1a/NR2B receptor function by D1R activation The above result reveals an important role of PSD-95 involved in D1R-modulated NR1a/NR2B receptor function. As we know, D1R activation can lead to an increase in cAMP formation and PKA activation. We next tested whether PKA is involved in D1R-enhanced NR1a/NR2B receptor-mediated Ca2+ influx in the cells which were co-expressed with PSD-95. After bath application of PKA selective inhibitor H89 (5 µmol/L), NR1a/NR2B receptor-mediated Ca2+ influx was significantly attenuated; an average 38.7%±4.1% of inhibition was observed (F1,2=66.40, n=13, P<0.01, compared to that in the absence of H89, Figure 5), indicating that NR1a/NR2B receptor-mediated Ca2+ influx is subjected to PKA regulation. Furthermore, H89 not only completely abolished the DA-enhanced Ca2+ influx mediated by NMDA agonists, but also resulted in an additional inhibition while DA was presented. To check if H89-mediated inhibition is reversible, H89-treated cells were washed out for 10 min prior to the reapplication of Glu/Gly. However, NR1a/NR2B receptor-mediated Ca2+ influx was unable to recover from the inhibition (Figure 5A).
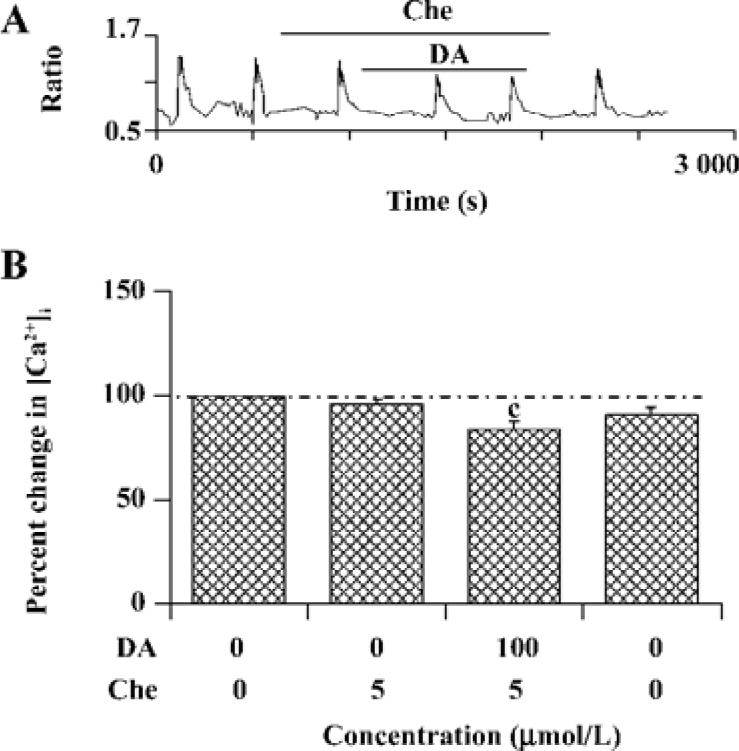
PKC is involved in the modulation of NR1a/NR2B receptor function induced by D1R activation As D1R may also activate PKC via a phospholipase C (PLC)-mediated mechanism and NMDAR was reported to be phosphorylated by PKC, the role of PKC was explored. NR1a/NR2B receptor-mediated Ca2+ influx was not altered by selective PKC inhibitor chelerythrine (4.0%±2.4% of inhibition, n=15, Figure 6). However, chelerythrine (5 µmol/L) indeed attenuated the DA-enhanced NR1a/NR2B receptor-mediated Ca2+ influx (15.8%±3.4%, F1,27=24.89, n=15, P<0.01) with less potency than that of H89. Thus, it indicates that PKC also contributed to the D1R modulation of NR1a/NR2B receptor function.
DiscussionOther Section
The present study demonstrates that in HEK293 cells co-transfected with the D1R and NR1a/NR2B receptor, D1R activation enhances NR1a/NR2B-mediated Ca2+ influx only in the presence of PSD-95. Moreover, when PKA or PKC activity was inhibited, the D1R-modulated NR1a/NR2B receptor function was also significantly attenuated. To our knowledge, this is the first evidence that D1/NR1a/NR2B receptor interaction depends on PSD-95.
PSD-95 is a scaffold protein abundant in PSD. PSD-95 can recruit signaling proteins and mediate assembly with other components of the PSD. It is known that PSD-95 plays an important role in mediating neurotransmitter receptor functions such as NMDA and 5-HT2A receptors[13–15]. The present result indicates that D1R activation fails to modulate NR1a/NR2B receptor-mediated Ca2+ influx unless PSD-95 is co-expressed with the D1R and the NR1a/NR2B receptor. Thus, it is clear that PSD-95 is required for D1R-modulated NR1a/NR2B receptor function. Other studies also support the important role of scaffold proteins in the regulation of NMDAR function in transfected HEK293 cells. For instance, the PKA-modulated NMDA current was significantly enhanced while the cells were co-expressed with yotiao, another scaffold protein enriched in the PSD[17]. We here found that the physical interaction between PSD-95 and D1R was essential for the functional expression of D1R-modulated NMDAR function. It appears that PSD-95 acts as a core component in recruiting D1R, NMDAR, and other signal transduction molecules to form a multiple protein complex that allows the interaction between D1R and NMDAR. Indeed, it has been suggested that PSD-95 facilitates the dynamic regulation of phosphoprotein (such as PKA or PKC) and sequentially attaches them to the substrate (for example, the receptors). In this manner, the signals can be efficiently transduced from one kinase to the next[18]. It remains unknown how D1R and PSD-95 interact, and furthermore, how this physical interaction regulates NR1a/NR2B receptor function.
Both PKA and PKC are found to be involved in the D1R modulation of NR1a/NR2B receptor function in the presence of PSD-95 in our system. Previous findings also suggested that an anchored pool of PKA was required for the augmentation of NMDAR-induced currents in HEK293 cells[17]. In neurons, the activation of the D1R has been shown to enhance the NMDA current via PKA- and PKC-dependent mechanisms[9,10]. It has also been reported that PKA, but not PKC, phosphorylates NMDAR or receptor-associated proteins, thereby inducing a conformational change of the receptor[19]. It appears that the role of PKA and PKC in D1R-modulated NMDAR function varied according to the cell system employed[19].
A previous report of HEK293 cells co-expressed with D1R and NMDAR indicated that the activation of D1R could lead to an inhibition of the NMDAR current[7]. It is worthy to note that the PKA and PKC inhibitors were used throughout the experiments in this study in order to exclude the potential post-translational effect of D1R-stimulated PKA or PKC on NMDAR function. Thus, the observed inhibitory effect of D1R activation on the NMDA current is more likely a result of the conformation change due to the direct physical association between the two receptors in their system. Indeed, it appears that there are two potential mechanisms for D1R-modulated NMDAR function. The confirmation change resulted from the physical interaction between the two receptors inhibiting the NMDAR function[7], whereas post-translational modification such as PKA or PKC-mediated phosphorylation enhanced NMDAR function[9,10]. The present data demonstrated that D1R- enhanced NR1a/NR2B Ca2+ influx was observed only in the presence of PSD-95, which was indeed subjected to the regulation of PKA or PKC, indicating that post-translational mechanisms play an essential role in D1R-modulated NMDAR function.
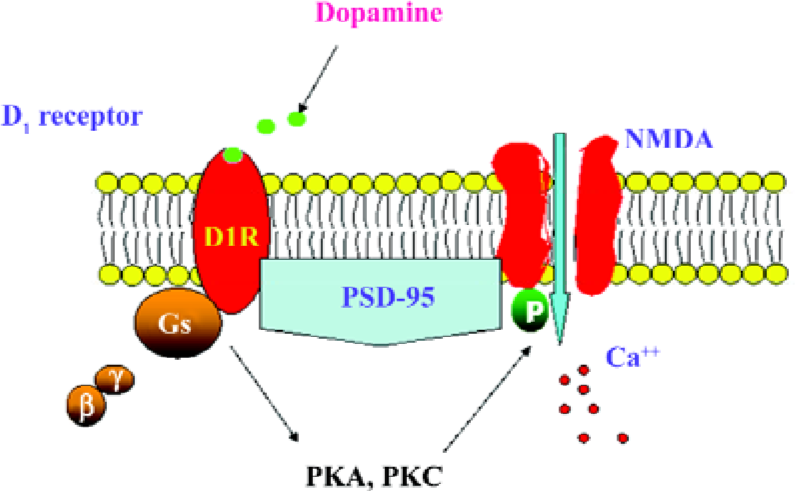
In summary, the present results demonstrate that the D1R activation of NR1a/NR2B receptor function requires the presence of PSD-95. The activation of D1R enhances NR1a/NR2B receptor-mediated Ca2+ influx and is dependent on PKA and PKC (Figure 7). It appears that PSD-95 is a core molecule in recruiting the two receptors to form a functional complex.
ReferencesOther Section
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev 1999;51:7-61.
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 2001;11:327-35.
- Gracy KN, Pickel VM. Ultrastructural immunocytochemical localization of the N-methyl-D-aspartate receptor and tyrosine hydroxylase in the shell of the rat nucleus accumbens. Brain Res 1996;739:169-81.
- Cepeda C, Levine MS. Dopamine and N-methyl-D-aspartate receptor interactions in the neostriatum. Dev Neurosci 1998;20:1-18.
- Sesack SR, Carr DB, Omelchenko N, Pinto A. Anatomical substrates for glutamate-dopamine interactions: evidence for specificity of connections and extrasynaptic actions. Ann N Y Acad Sci 2003;1003:36-52.
- Fiorentini C, Gardoni F, Spano P, Di Luca M, Missale C. Regulation of dopamine D1 receptor trafficking and desensitization by oligomerization with glutamate N-methyl-D-aspartate receptors. J Biol Chem 2003;278:20196-202.
- Lee FJ, Xue S, Pei L, Vukusic B, Chery N, Wang Y, et al. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell 2002;111:219-30.
- Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci 1988;18:10297-303.
- Wang J, O’Donnell P. D. (1) dopamine receptors potentiate NMDA-mediated excitability increase in layer V prefrontal cortical pyramidal neurons. Cereb Cortex 2001;11:452-62.
- Chen G., Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci USA 2004;101:2596-600.
- Kennedy MB. Signal-processing machines at the postsynaptic density. Science 2000;290:750-4.
- McGee AW, Dakoji SR, Olsen O, Bredt DS, Lim WA, Prehoda KE. Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol Cell 2001;8:1291-301.
- Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci 1996;16:2157-63.
- Xie Z, Gray JA, Compton-Toth BA, Roth BL. A direct interaction of PSD-95 with 5-HT2A serotonin receptors regulates receptor trafficking and signal transduction. J Biol Chem 2003;278:21901-8.
- Swayze RD, Lise MF, Levinson JN, Phillips A, El-Husseini A. Modulation of dopamine mediated phosphorylation of AMPA receptors by PSD-95 and AKAP79/150. Neuropharm 2004;47:764-78.
- Barton AC, Sibley DR. Agonist-induced desensitization of D1-dopamine receptors linked to adenylyl cyclase activity in cultured NS20Y neuroblastoma cells. Mol Pharmacol 1990;38:531-41.
- Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, et al. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science 1999;285:93-6.
- Posas F, Saito H. Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science 1997;276:1702-5.
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, et al. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci 2006;9:501-10.