
Redispersible, dry emulsion of lovastatin protects against intestinal metabolism and improves bioavailability1
Introduction
Lovastatin (Lov; Figure 1A), a cholesterol-lowering agent, is an inactive lactone that can be converted to the corresponding active β-hydroxy acid form (lovastatin acid [Lova]; Figure 1B). It is a potent inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase. The enzyme catalyzes the conversion of HMG-CoA to mevalonate in the liver as a specific target organ, which is an early and rate-limiting step in cholesterol biosynthesis[1]. Lov lactone has favorable hepatic selectivity; however, the poor absorptive characteristics and high prehepatic metabolism of Lov limits the extent of liver extraction and results in decreased clinical efficacy[2]. The incomplete dissolution in gastrointestinal fluids (solubility 0.4 μg·mL–1) and low intestinal membrane permeability (LogP 4.07) could account for the low and erratic absorptive extent (23%−30%) upon oral administration. In addition, Lov is prone to extensive gut wall metabolism with cytochrome P450 3A as the major enzyme, and transmembrane efflux by P-glycoprotein located in the apical enterocyte membrane of the small intestine[3]. The synergistic effects of the 2 proteins suggest that intestinal metabolism plays a role in prehepatic clearance[4]. Thus increasing the dissolution rate and decreasing prehepatic metabolism may be useful for enhancing the amount of Lov in oral absorption.

A variety of formulation principles, such as micronization, solid dispersions[5], the cyclodextrin (CD) complex[6], and solid lipid nanoparticles[7] have been investigated in order to increase the dissolution rate of the poorly water-soluble drug, but these techniques could not reduce intestinal extraction, which is an important factor determining the absorptive efficiency of Lov. Lipid-based formulations, especially emulsified formulations, have shown their utility in enhancing the absorption of poorly water-soluble drugs based on increasing drug dissolution in the gastrointestinal tract[8]. In addition, certain lipid-based formulation ingredients, such as Tween 80[9,10] and Cremophor EL[11], have potential inhibitory effects on intestinal cytochrome P450 or P-glycoprotein. The oil/water emulsion composed of the active formulation ingredients would be a suitable formulation for Lov to reduce intestinal extraction, albeit with a relatively large volume and physical instability. Notably, dry emulsion (DE) has been suggested as one way to circumvent the disadvantage of conventional liquid lipid-based formulations[12,13] and has been successfully applied as a potential oral drug delivery system for improving solubility and dissolution of poorly soluble drugs[14,15]. However, no current information is available for protecting drug substances against intestinal metabolism by DE.
The objective of the present study was to develop a redispersible spray-dried emulsion capable of protecting against intestinal metabolism and enhancing the oral absorptive efficiency of Lov. The formulation was characterized for its dissolution rate in vitro, as well as its potential to prevent metabolism in microsomes and the gut wall in male Sprague–Dawley (SD) rats. We also evaluated the bioavailability of the redispersible, DE of Lov (Lov-DE) compared to the control formulations [Lov suspension (Lov-Sus) and Lov–CD complex].
Materials and methods
Chemicals Lov, Lova, and simvastatin hydroxyl acid were purchased from Zhejiang Ruibang Laboratories (Zhejiang, China). Phosal 53 MCT (consisting of phospholipids and medium chain triglyceride) was purchased from Lipoid GmbH (Rhine, Germany). Tween 80 was purchased from Sinopharm Chemical Reagent (Shanghai, China). Starch sodium octenyl succinate was a gift from National Starch (Berkeley, California, USA). Hydroxypropyl (HP)–β-CD was purchased from Shangdong Xinda (Shangdong, China). Solid-phase extraction cartridges were obtained from Waters (Oasis HLB; Manchester, UK). Double-distilled quality water was used. All other reagents were of analytical grade or higher.
Animals Male SD rats were obtained from the Medical Animal Test Center of Shanghai Institute of Materia Medica (Shanghai, China). All of the experiments were performed according to the Shanghai Institute of Materia Medica guidelines of experimental animal care. The rats fasted for 12 h prior to the experiment and had free access to water.
Preparation of formulations Lov-DE was prepared as follows: Lov (0.5 g) was dissolved in a mixture of oil phase Phosal 53 MCT (9 g) and emulsifying agent Tween 80 (1 g). The matrix material starch sodium octenyl succinate (10 g) was dissolved in 500 mL water as the aqueous phase. Both phases were brought to 60 ºC separately in a water bath, then prehomogenized to form coarse emulsion at 1350×g for 5 min using Ultra Turrax (T25; IKA, Staufen, Germany). The coarse emulsion was homogenized at 80 MPa 8 times in a high-pressure homogenizer (Panda2K; GEA, Segrate, Italy). The resulting submicron emulsion was spray dried immediately in a Mobile Minor spray dryer (Niro, Soeborg, Denmark) under the following conditions: inlet temperature, 180 ºC; outlet temperature, 70 ºC; and feeding rate of the emulsion, 10 mL·min-1.
Lov and methylcellulose mucilage (0.5% w/v) were ground in a mortar to obtain a 4 mg·mL-1 Lov-Sus and were ultrasonicated for 2 min. The average particle size (n=100) was 4.76±1.87 μm measured by an optical microscope (CKX41; Olympus, Tokyo, Japan).
The Lov-CD complex was prepared by adding Lov to the ethanol solution of HP–β-CD (1:2 molar ratio) and stirred for 15 min. The Lov–CD complex was obtained by evaporating the ethanol using rotavapor (Buchi, Flawil, Switzerland). The inclusion of Lov in CD was confirmed by differential scanning calorimetry (STAR, Mettler–Toledo, Barcelona, Spain).
Characterization of formulations The particle size distribution of Lov-DE was measured by a laser particle size analyzer (Mastersizer 2000; Malvern, Worcestershire, UK) after dispersing the DE powder in air flow. The particle size was expressed as volume mean diameter. Droplet size and the polydispersity index of the emulsions before spray drying and after reconstituting Lov-DE were determined by photon correlation spectroscopy using Nicomp 380 ZLS (Particles Sizing Systems, Santa Barbara, California, USA). The droplet size was recorded as intensity distributions.
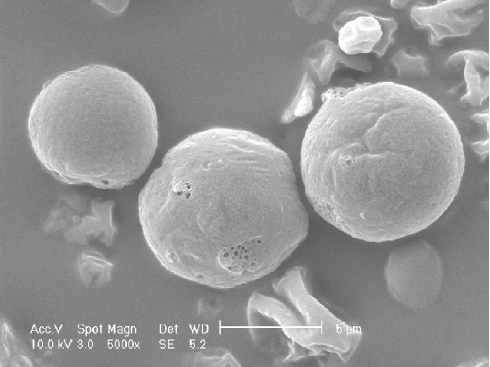
The visualization of morphology was achieved by scanning electron microscopy (SEM). Scanning electron micrographs were taken using a Philips XL 30 (Philips, Eindhoven, Netherlands). Samples were fixed on an aluminum stub with conductive double-sided adhesive tape and sputter coated with gold in an argon atmosphere (50 Pa) at 50 mA for 50 s.
The total drug content in the DE was obtained using HPLC (1100 series; Agilent, Santa Clara, California, USA) after dissolving the colloidal dispersions by methanol. The encapsulation efficiency (EE) of Lov-DE was conducted by ultrafiltration. Free Lov (non-incorporated in the DE) in the ultrafiltrate was determined after separation of the reconstitution emulsion by ultrafiltration centrifugation technique (Microcon YM-10, 10 000 MW; Millipore, Billerica, USA).
The HPLC wavelength was set to λ/nm=237. An Inertsil ODS3-C18 column (4.6×150 mm, 5 μm; GL Science, Tokyo, Japan) was used. The mobile phase consisted of acetonitrile: water:glacial acetic acid (70:30:0.5, v/v/v). The EE was calculated by the following equation:
EE (%)=
where WT and WF are the weight of the total drug in DE and free drug in the ultrafiltrate detected after centrifugation.
The conversion of Lov from its lactone form to its corresponding hydroxy acid form in different formulations was determined using HPLC. The reconstituted properties of Lov-DE were determined at the designated time intervals after its storage in a sealed bottle at room temperature. The droplet size of reconstituted DE was measured for 3 months.
In vitro release study The in vitro release of Lov from various formulations was studied using the bulk equilibrium reverse dialysis bag technique. The dialysis bags (cellulosic membranes with molecular weight cut-offs of 12 000–14000 Da; Millipore, USA) were placed in a RCZ-8B dissolution tester (Tiandatianfa, Tianjin, China) containing 900 mL dissolution media (phosphate-buffered solution [PBS] [pH 4.0] with 0.5% SDS)[1 ]. The temperature was maintained at 37 ºC, and the paddle rotation speed was set to 50 rpm. Formulations (equivalent to 0.5 mg Lov) were directly placed into the stirred sink solution in which numerous dialysis sacs containing 2 mL of the same sink solution were immersed in advance. The dialysis sacs were equilibrated with the sink solutions for approximately 30 min prior to experimentation. At predetermined time intervals, samples were withdrawn from the dialysis bags and filtered through a membrane filter (0.22 μm), and the contents were analyzed by the HPLC method.
Microsomal metabolism study Liver microsomes were prepared from untreated male SD rats weighing 300-350 g[3]. Microsomal protein concentrations were measured by the Lowry method using bovine serum albumin as the standard. The standard incubation mixture contained 100 μmol potassium phosphate buffer (pH 6.8), 0.5 μmol NADPH, 1 mg microsomal protein, and 100 nmol Lov formulations in a final volume of 1 mL. The reaction mixture was incubated for 15 min at 37 ºC in a water bath. At the end of the incubation period, the reaction was stopped by the addition of 1 mL acetone, and the unreacted substrates and metabolites were extracted using 2 mL ethyl acetate. After centrifugation, the organic layer was carefully removed and evaporated under a nitrogen stream at 40 ºC. Concentrated extracts were stored at -20 ºC until the HPLC analysis. Blank incubations consisted of no NADPH.
Intestinal metabolism study Lov-DE, Lov-Sus, and Lov-CD were administered to the rats by oral gavage at a dose of 20 mg·kg-1[2]. The formulations were dispersed in water prior to dosing. The rats were anesthetized with an intraperitoneal injection of ethylcarbamate at a dose of 1 g·kg-1, and blood samples were simultaneously withdrawn from the hepatic portal vein at 30, 60, or 120 min postadministration into the heparinized tubes.
Pharmacokinetics study Lov formulations were administered to the rats by oral gavage at a dose of 20 mg·kg-1, similar to the intestinal metabolism study. Blood samples were withdrawn by retro-orbital venous plexus puncturation into the heparinized tubes at designated time intervals (0, 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, and 8 h). After centrifugation, the plasma obtained was stored at –20 ºC until analysis.
Analysis of Lov in rat plasma Frozen plasma samples were thawed at room temperature just prior to the extraction procedure and agitated, with the whole procedure performed in the dark. In total, 20 μL of internal standard (simvastatin acid, 10 μg·mL-1 in acetonitrile:water [70:30, v/v]) was added into 200 μL plasma and mixed for 30 s. The mixture was transferred into a Waters Oasis HLB solid-phase extraction cartridge, which had been activated with 1 mL methanol and 1 mL distilled water[3]. Samples were obtained by washing the extraction cartridge with 1 mL of 5% methanol twice. The samples were eluted using 1 mL methanol. This fraction was evaporated under a nitrogen stream at 40 ºC. The samples were reconstituted with 100 μL of mobile phase, and 50 μL of the eluted fraction was injected into the HPLC column.
Statistical analysis The maximal plasma concentration of Lov (Cmax) and the time to reach Cmax (Tmax) were directly obtained from plasma data. The area under the drug concentration–time curve (AUC) and other pharmacokinetic parameters were analyzed using Drug and statistics (DAS) 2.0 software (Anhui, China). Data from different formulations were analyzed by Student’s t-test. The result was presented as mean±SD, and the differences were considered significant when P<0.05 or P<0.01.
Results
Preparation of DE To select an appropriate oil phase, the solubility of Lov in various oils (eg soybean oil, castor oil, medium chain triglyceride, Phosal 53 MCT) was determined. Lov had the highest solubility in Phosal 53 MCT (58.7 mg·g-1), which is a biocompatible ingredient consisting of phospholipids and MCT. Therefore Phosal 53 MCT was chosen as the oil phase. The emulsifying agent Tween 80 has potential inhibitory effects on intestinal metabolism. Starch sodium octenyl succinate is a safe and good reconstitution characteristics matrix[4] and also a stabilizer which makes the submicron emulsions more stable, therefore this material was applied as matrix in the oral lipid formulation for the first time. The process of preparing and spray-drying submicron emulsion was selected for the optimal DE formulation. The spray-drying yield of the DE was 74.3%±3.2%.
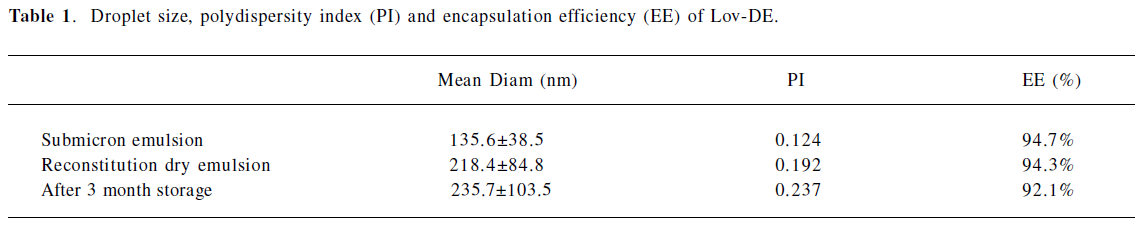
Characterization of DE The particle size distribution of Lov-DE measured in compressed air indicated that spray-dried powders had the middle value below 5 μm and exhibited tight particle size distribution as reflected by the 10% and 90% values of 2.937 and 8.254 μm, respectively. The outer macroscopic morphology of Lov-DE was seen as a well-separated particle with a smooth surface, as revealed by SEM scanning (Figure 2). The droplet size analysis (Table 1) showed that the size distribution intensity values for the submicron emulsion before spray drying and the reconstituted DE were 135.6±38.5 and 218.4±84.8 nm, respectively, which showed good reconstitution properties of Lov-DE. The Lov content in the DE was determined to be 0.97% (w/w), close to the theoretical drug content of 0.99% (w/w), with an encapsulation efficiency of 94.3%. The DE exhibited good physical stability after 3 months of storage, as indicated by reconstitution properties, including intensity distribution values of 235.7±103.5 nm and encapsulation efficiency of 92.1%. The data also showed that the Lov lactone form was hardly converted to its hydroxyl acid form in the solid state (2.7%), but was almost half converted to its hydroxyl acid form in the liquid state after 3 months (45.5%).
Full table
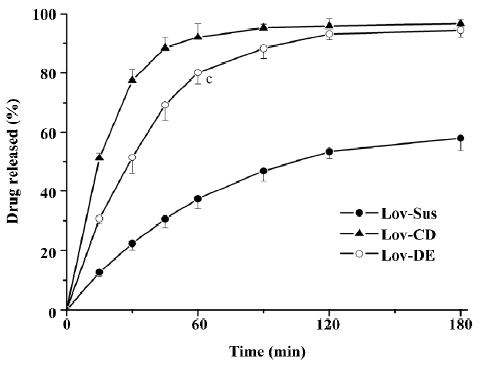
Drug release in vitro In order to evaluate the dissolution effect of the prepared formulations, Lov released in vitro from the formulations was investigated over 3 h. The percentages of Lov released from Lov-DE, Lov-CD, and Lov-Sus were 80.1%, 92.1%, and 37.3% at 60 min, respectively. Therefore, the amount of Lov released from DE into the medium was similar to that from HP-β-CD, but greater than that from the suspensions (Figure 3). This suggested that the Lov-incorporated, lipid-based formulation and complex with HP-β-CD improved the dissolution characteristics obviously.
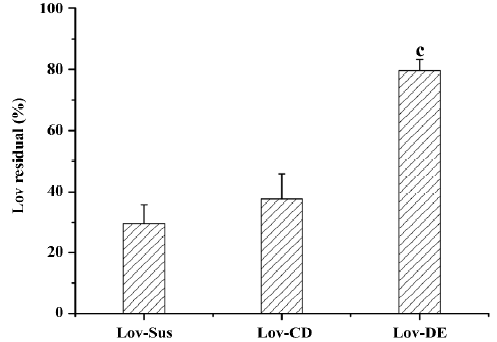
Microsomal metabolism study The metabolism assay carried out in rat microsomes evaluated the protective effects of the different dosage forms. After incubation with microsomes (1 mg·mL-1) for 15 min, the parent drug amount of the 3 formulations was measured. The protective effects of the formulation could be defined as the percentage of the residual parent drug which had not been metabolized by microsomal enzymes. The residual Lov percentages of the tested formulations were 29.6%, 27.6%, and 79.6% for Lov-Sus, Lov-CD, and Lov-DE, respectively. Lipid-based formulation exhibited significant protective effects against enzymatic attack in rat microsomes compared with Lov-Sus and Lov-CD (P<0.01; Figure 4).
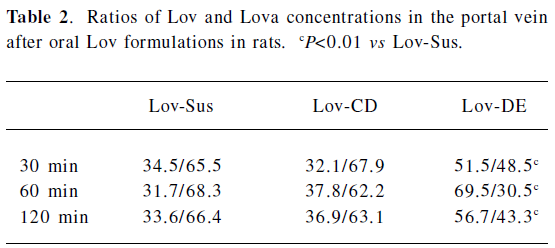
Animal study Lov and Lova concentrations were determined in the blood samples of the rat portal vein. Drug concentrations and the ratios of Lov/Lova concentrations in the plasma are shown in Figure 5 and Table 2. The results showed that drug absorption (total amount of Lov and Lova) was significantly enhanced by both of formulations compared with the control suspension at 3 tested points (P<0.05). However, the ratios of Lov/Lov concentrations showed that Lov-DE and Lov-CD had different characteristics for increasing absorption. The Lov lactone percentage, administered by Lov-DE, was significantly greater than that of Lov-Sus (P<0.01), and Lov-CD showed no statistical difference to Lov-Sus.
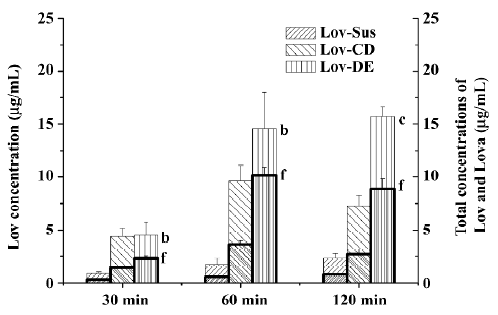
Full table
The Cmax and AUC0–8 h of Lov and Lova after the oral administration of DE were 2.3- and 1.8-fold higher than those of the suspension, and were comparable to those with HP-β-CD administration (Figure 6 and Table 3). Similar to the result of in vitro release, both Lov-DE and Lov-CD exhibited a shorter time to reach Tmax and higher Cmax. Compared with Lov-CD, the Cmax and the AUC of Lov-DE were increased more significantly.
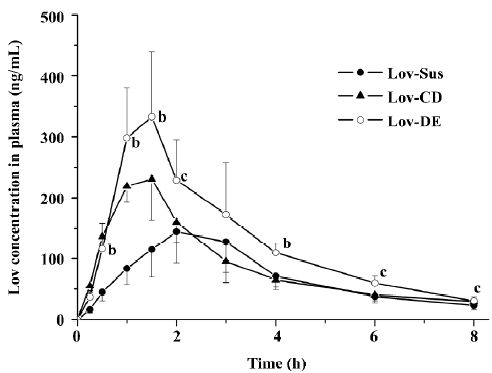
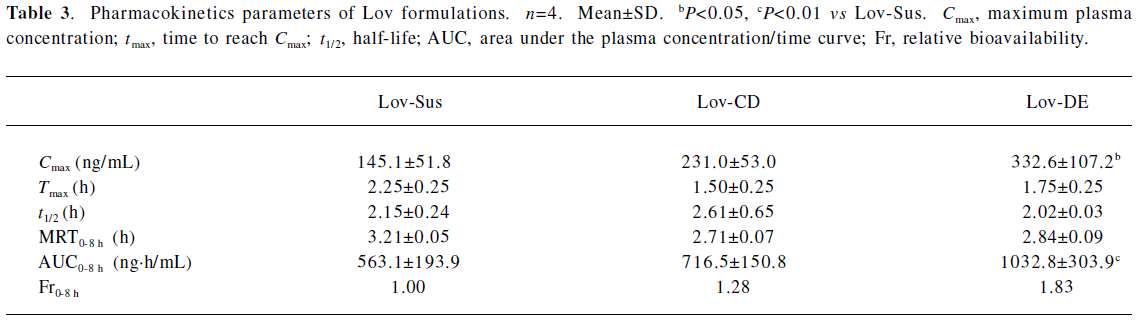
Full table
Discussion
In the present study, the potential of DE as a lipid-based delivery system for Lov was evaluated with respect to redispersibility, physical stability, in vitro release characteristics, and protective effects against enzymatic attack, as well as in vivo absorption. The DE was prepared by spray drying the o/w submicron emulsion of Lov.
The DE developed in this study could be dispersed well upon reconstitution with water, and the resulting droplet distribution of the emulsion was homogeneous with the size of only 218 nm. Our data indicated that the DE-entrapped Lov could retain good accommodation of the drug without any conversions from the lactone form to the hydroxyl acid form for at least 3 months. The results suggested that the physical stability of the Lov-DE was improved compared to that of the conventional o/w emulsion.
The Lov-DE could protect Lov from enzymatic attack, as indicated by the microsome incubation test (79.6% vs 29.6% of parent drug residue after 15 min compared to Lov suspension). However, Lov-CD did not display similar effects. The most accountable reason for this phenomenon was the entrapment of Lov in the lipid-based formulation. Lipid ingredients like a protective cover which decreased the possibility of the contacts between enzymes and Lov. Another reason may be the inhibition of microsome metabolism in the presence of the surface-active ingredients, such as Tween 80 in lipid-based formulations[5]. Lov in the CD complex remained exposed to enzymes. Meanwhile, CD was not an inhibitor for CYP3A, so Lov-CD had no protective effects on Lov.
The total amount of Lov absorption which passed through the intestinal wall into the hepatic portal vein was enhanced by improved dissolution characteristics. For instance, the percentages of Lov released from Lov-DE, Lov-CD, and Lov-Sus were 80.1%, 92.1%, and 37.3% at 60 min in vitro, respectively, and the total amount of Lov were 14.57, 9.63, and 1.74 μg·mL–1 in the hepatic portal vein, correspondingly. Lov-DE allowed Lov to remain in a dissolved state within a colloidal dispersion throughout its transition through the gastrointestinal tract[6]. Meanwhile, the presence of intermolecular hydrogen bonds between Lov and HP-β-CD in the inclusion complex resulted in Lov maintaining its molecular form in Lov-CD[6]. Therefore, Lov-DE and Lov-CD could improve dissolution and absorption obviously. However, the intestinal metabolism results suggested that the metabolic features of Lov were different from Lov-DE and Lov-CD, even though Lov absorption was enhanced in both formulations. For example, Lov lactone in Lov-DE was 69.5% (P<0.01) at 60 min, while it was 37.8% in Lov-CD (P>0.05) in comparison, and 31.7% in Lov-Sus. These interesting results coincided with those of the microsome metabolism assay in vitro. Additionally, it was reported that Lov lactone had more favorable hepatic selectivity than Lova[2], so the drug extracted by the liver may be enhanced considerably as a result of high levels of Lov lactone in the hepatic portal vein. This hypothesis was supported by the data analyzed by DAS 2.0, which showed that Lov in the liver of Lov-DE was 7.1-fold higher than that of Lov-Sus in the first 2 h after administration. Therefore, it is reasonable to conclude that a higher hepatic drug concentration of Lov-DE may lead to better therapeutic effects.
The pharmacokinetic results indicated that Lov-DE significantly improved the bioavailability of Lov compared with Lov-Sus and Lov-CD. This may due to the fact that submicron emulsion markedly improved dissolution and reduced metabolism in the gut wall, and therefore, enhanced the absorption of Lov in vivo. Compared with Lov concentrations in the portal vein and systemic circulation, it was found that the enhanced absorption of Lov in the liver was not necessarily proportional to the elevated plasma AUC. Lov-DE showed a significantly higher amount of Lov in the liver (7.1-fold) and was slightly higher in the plasma (1.8-fold) compared with Lov-Sus, which means that Lov-DE had more favorable hepatic selectivity. Lov specifically targets the liver, and like other inhibitors of HMG-CoA reductase, it may increase the risk of myopathy/rhabdomyolysis when the plasma concentration of Lov is too high[7]. Therefore, organic selectivity is more important than bioavailability for Lov. In order to clarify this issue, the pharmacodynamics of Lov-DE should be further investigated. Considering the compliance of patients, DE may be more suitable for long-term administrated drugs whose absorptions are not only limited by dissolution/solubility, but also by intestinal metabolic instability[16-22].
Solid state emulsions would be a convenient way to avoid the thermodynamic and physicochemical instability of compounds in liquid media. DE developed in this study reconstituted efficiently with improved physical stability. Moreover, DE could protect Lov from intestinal metabolism and enhance the oral absorption efficiency of Lov lactone in the liver significantly. In addition, compared with Lov-Sus, DE also improved the bioavailability of Lov. Our work suggests that DE is a potential drug delivery system for other lipophilic drugs with similar physicochemical characteristics, such as poor solubility and intestinal metabolic instability upon oral administration.
References
- Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, et al. Mevinolin: a highly potent competitive inhibitor of hydroxy-methylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci USA 1980;77:3957-61.
- Reinoso RF, Sánchez Navarro A, García MJ, Prous JR, et al. Preclinical pharmacokinetics of statins. Methods Find Exp Clin Pharmacol 2002;24:593-613.
- Jacobsen W, Kirchner G, Hallensleben K, Mancinelli L, Deters M, Hackbarth I, et al. Small intestinal metabolism of the 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor lovastatin and comparison with pravastatin. J Pharmacol Exp Ther 1999;291:131-9.
- Zhang Y, Benet LZ. The gut as a barrier to drug absorption: combined role of cytochrome P450 3A and P-glycoprotein. Clin Pharmacokinet 2001;40:159-68.
- Patel RP, Patel MM. Physicochemical characterization and dissolution study of solid dispersions of Lovastatin with polyethylene glycol 4000 and polyvinylpyrrolidone K30. Pharm Dev Technol 2007;12:21-33.
- Jun SW, Kim MS, Kim JS, Park HJ, Lee S, Woo JS, et al. Preparation and characterization of simvastatin/hydroxypropyl-beta-cyclodextrin inclusion complex using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm 2007;66:413-21.
- Suresh G, Manjunath K, Venkateswarlu V, Satyanarayana V. Preparation, characterization, and in vitro and in vivo evaluation of lovastatin solid lipid nanoparticles. AAPS PharmSciTech 2007;8:E162-E170.
- Charman WN, Porter CJH, Mithani S, Dressman JB. Physicochemical and physiological mechanisms for the effects of food on drug absorption: the role of lipids and pH. J Pharm Sci 1997;86:269-82.
- Daher CF, Baroody GM, Howland RJ. Effect of a surfactant, Tween 80, on the formation and secretion of chylomicrons in the rat. Food Chem Toxicol 2003;41:575-82.
- Wang SW, Monagle J, McNulty C, Putnam D, Chen H. Determination of P-glycoprotein inhibition by excipients and their combinations using an integrated high-throughput process. J Pharm Sci 2004;93:2755-67.
- Rege BD, Kao JP, Polli JE. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur J Pharm Sci 2002;16:237-46.
- Dollo G, Le Corre P, Guérin A, Chevanne F, Burgot JL, Leverge R. Spray-dried redispersible oil-in-water emulsion to improve oral bioavailability of poorly soluble drugs. Eur J Pharm Sci 2003;19:273-80.
- Tue H, Per H, Morten R, Kirsten S. In vivo evaluation of tablets and capsules containings spray-dried o/w emulsions for oral delivery of poorly soluble drugs. Int J Pharm 2005;293:203-11.
- Dixit RP, Nagarsenker MS. Dry adsorbed emulsion of simvastatin: optimization and in vivo advantage. Pharm Dev Technol 2007;12:495-504.
- Chambin O, Be’rard V, Rochat-Gonthier MH, Pourcelot Y. Dry adsorbed emulsion: 2. Dissolution behaviour of an intricate formulation. Int J Pharm 2002;235:169-78.
- Chen CM, Chou J, Wong D. Improved HMG-CoA reductase inhibitor extended release formulation. US patent 5 916 595. 1998 Dec 04.
- Duggan DE, Bayne WF, Halpin RA, Duncan CA, Schwartz MS, et al. The physiological disposition of lovastatin. Drug Metab Dispos 1989;17:166-73.
- Stubbs RJ, Schwartz M, Bayne WF. Determination of mevinolin and mevinolinic acid in plasma and bile by reversed-phase high-performance liquid chromatography. J Chromatogr 1986;383:438-43.
- World Health Organization. Toxicological evaluation of certain food additives (Starch sodium octenyl succinate). WHO Food Additives Series 1982;17:541.
- Gershanik T, Benita S. Self-dispersing lipid formulations for improving Coral absorption of lipophilic drugs. Eur J Pharm Biopharm 2000;50:179-88.
- Pouton CW. Lipid formulations for oral administration of drugs non-emulsifying, self-emulsifying and self-microemulsifying drug delivery systems. Eur J Pharm Sci 2000;11 Suppl 2:S93-8.
- Mountfield RJ, Senepin S, Schleimer M, Walter I, Bittner B. Potential inhibitory effects of formulation ingredients on intestinal cytochrome P450. Int J Pharm 2000;211:89-92.