
Human urine extract CDA-2 induces apoptosis of myelodysplastic syndrome-derived MUTZ-1 cells through the PI3K/Akt signaling pathway in a caspase-3-dependent manner1
Introduction
Myelodysplastic syndromes (MDS) are clonal disorders characterized by trilineage defects in hematopoiesis[1,2], including erythrocytic, granulocytic, and megakaryocytic lineages. Although clonal, MDS is sometimes considered a premalignant condition that often progresses to acute myeloid leukemia (AML). The classification of MDS into low-grade refractory anemia (relatively long survival), intermediate refractory cytopenia with multilineage dysplasia, and high-grade refractory anemia with excess blasts depends on percent blasts, karyotype, and peripheral cytopenias[3]. Intermediate and high-grade cases manifest multiple abnormalities typically including neutropenia and granulocytic dysplasia that frequently progress to AML with an especially poor prognosis[4]. Finding new agents that might overcome intermediate and high-grade MDS has thus been actively pursued.
Urine and urine extracts for therapeutic purposes have been used for centuries[5–7]. Cell differentiation agent 2 (CDA-2) is a mixture product, isolated from healthy human urine in China. Previous reports have suggested that CDA-2 acts as a novel anticancer agent having multiple biological targets in antiproliferation, apoptosis, differentiation, and gene regulation in several solid tumors[7]. Multiple active components, such as peptides (MW 400-2800), organic acids, phenylacetate, and pigments with different mechanisms of action act concurrently to contribute to the anticancer effect of CDA-2[8]. The State Drug Administration (SDA) of China approved the use of CDA-2 as an anticancer drug in solid tumors in August 2004.
Most MDS patients are elderly, and all of them have cytopenias. CDA-2 has relatively mild medullary and extra-medullary toxicities, which makes it a suitable candidate for the treatment of MDS. However, there has been no report that clearly describes the main mechanisms of CDA-2 in MDS cells.
In the present study, we evaluated both in vitro and in vivo anticancer activities and the mechanism of action of CDA-2 in MDS models in order to provide experimental therapeutic data to expand its indications in clinical trials.
Materials and methods
Preparations of the human urine extract CDA-2 was kindly supplied by Ever Life Pharmaceutical (Hefei, Anhui, China). The human urine was acidified during collection and passed through an ultrafiltration process to remove molecules with molecular weights greater than 10 000 daltons. The filtrate was then passed through a chromatographic column and eluted by ethanol. The colored ethanolic fraction was collected and evaporated under vacuum. The dried extract was reconstituted with distilled water to make a 300 g/L stock solution and stored at 4 ºC. The compositions of CDA-2 included organic acids, peptides, pigments, and phenylacetylglutamine[8,9].
Cell culture and agents The MUTZ-1 cell line was kindly provided by Prof Zhen-bo HU from Chicago University (Chicago, IL, USA). It was established from the peripheral blood of a 5-year-old girl with MDS French-American-British (FAB) subtype refractory anemia with excess of blasts (RAEB)]. The MDS arose from a pre-existing Fanconi anemia and progressed quickly to an acute myeloid leukemia (FAB M2)[10]. The MUTZ-1 cells showed cytogenetic alterations associated with MDS: del (5) (q13/33) and contained the mutant P53 gene[11]. The MUTZ-1 cell line, acute myelocytic leukemia cell lines (HL-60, Kasumi-1, KG-1, U937, and NB4), chromic myeloid leukemia cell line (K562), Burkitt’s lymphoma cell line (Raji), and T-cell acute lymphoblastic leukemia cell line (Jurkat and Molt-4) were all maintained in RPMI-1640 (Gibco-BRL, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal calf serum (FBS; Gibco-BRL, USA), 0.2 g/L streptomycin/penicillin, and 0.1% (w/v) L-glutamine (Gibco-BRL, Grand Island, NY, USA) in a 5% humidified CO2 atmosphere at 37 ºC. Blood samples collected from healthy volunteers were processed by Ficoll Hypaque gradient to obtain normal peripheral blood mononuclear cells (PBMC). The caspase-3 inhibitor Z-DEVD-fmk (40 µmol/L) (BioVision, Mountain View, CA, USA) was added 1 h before the treatment with CDA-2. Phosphoinositide 3 kinase (PI3K) inhibitor LY294002 (30 µmol/L; Cell Signaling, Boston, MA, USA) was also added 1 h before the treatment with CDA-2.
Growth inhibition assay The inhibitory effects of CDA-2 on the proliferation of the MUTZ-1 cell line and human leukemia cell lines, as well as normal PBMC, were assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma, St Louis, MO, USA) assay. The cells were seeded at a density of 1×104/well in 96-well microtiter plates. The cells were treated with various concentrations of CDA-2 and incubated at 37 ºC in a 5% CO2 atmosphere for 24, 48, and 72 h. Afterwards, 20 µL of 5 g/L MTT stock solution was added to each well (final concentration: 0.5 g/L) for another 4 h of incubation (37 ºC, 5% CO2). After 4 h of incubation, 200 µL DMSO was added to each well, and the optical density was read at 570 nm. The sensitivity of the cells to CDA-2 was measured by IC50 (50% inhibitory concentration). Experimental conditions were tested in sextuplicate (6 wells of the 96-well plate per experimental condition). All the experiments were performed in triplicate.
Cell cycle analysis and apoptosis determination The MUTZ-1 cells (1×106) were washed twice in phosphate-buffered saline (PBS) and fixed with 70% ice-cold ethanol, then centrifuged and suspended in PBS containing 100 mg/L RNase A. After incubation for 30 min at 37 ºC, the samples were resuspended in 25 mg/L propidium iodide (PI; Sigma, USA). Apoptosis was measured by an Annexin-V–fluorescein-isothiocyanate apoptosis detection kit (BD Pharmingen, San Diego, CA, USA), according to the manufacturer’s instructions. Data acquisition and analyses were done on a Becton Dickinson (Franklin Lakes, NJ, USA) FACSCalibur using CellQuest software (Becton Dickinson, USA). Annexin-V binds to cells that express phosphatidylserine on the outer layer of the cell membrane, and PI stains the cellular DNA of cells with a compromised cell membrane. All the experiments were performed in triplicate.
Mitochondrial membrane potential measurement Alterations in the mitochondrial membrane potential (ΔΨm) were analyzed by flow cytometry using the ΔΨm-sensitive dye the lipophilic cation 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimi- dazolcarbocyanineiodide(JC-1) (Molecular Probes, Eugene, OR, USA). Briefly, following treatment, 2×106 cells were harvested, washed once, resuspended in PBS, and incubated with 1 µmol/L JC-1 at 37 ºC for 10 min. Stained cells were then washed once with PBS and analyzed by flow cytometry. A Becton Dickinson FACSCalibur was used to analyze a minimum of 1×104 cells per sample. JC-1 was a cationic dye that exhibited potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (525±10 nm) to red (610±10 nm). Data were evaluated using the CellQuest software package. Forward and side scatters were used to gate viable populations of cells. JC-1 monomers emit at 527 nm (FL-1 channel) and “J-aggregates” emit at 590 nm (FL-2 channel). All the experiments were performed in triplicate.
Western blot analysis and antibodies The MUTZ-1 cells were incubated with 2-8 g/L CDA-2 for 24 h. The cells (5×106) were harvested and lysed in 200 µL lysis buffer (0.5 mol/L Tris-HCl, pH 6.8, 2 mmol/L EDTA, 10% glycerol, 2% SDS, and 5% β-mercaptoethanol). The extracted total protein was loaded at 40 µg per lane on 12% Tris-glycine gels and then transferred to a polyvinylidine diflouride membrane (Millipore, Billerica, MA, USA). The membrane was blocked in 5% non-fat milk dissolved in Tris buffered saline with 0.1% Tween 20 (TBS-T), and subsequently probed with the primary antibody and a horseradish peroxidase-labeled secondary antibody. The bands were visualized using enhanced chemiluminescence Western blotting detection reagents (Amersham, Little Chalfont, UK). Primary antibodies included caspase-3, poly(ADP-ribose)polymerase (PARP), caspases-8 and -9, total Akt, Ser 473 phosphorylated Akt antibodies (p-Akt), cellular inhibitor of apoptosis protein 1(CIAP1), CIAP2, X-linked mammalian inhibitor of apoptosis protein (XIAP), NF-κB (P65), cyclin D1 (BioVision, Mountain View, CA, USA), PI3Kp110α, Bad, Ser 136 phosphorylated Bad (p-Bad), Bid, Bcl-2, Bax, survivin, Smac, total IκBα, Ser 32 phosphorylated IκBα, Ser 536 phosphorylated NF-κB (p-P65), FLICE-like inhibitory protein(FLIP), cyclin D2, cyclin D3, cyclin-dependent kinase (CDK)4, CDK6 (Cell Signaling, Danvers, MA, USA), actin, and lamin B (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Horseradish peroxidase-conjugated secondary antimouse and antirabbit antibodies were purchased from Santa Cruz Biotechnology (USA).
Nuclear and cytosolic fractionation The MUTZ-1 cells were treated with 2-8 g/L CDA-2 for 24 h. Then the cells (1×107) were harvested and incubated in 400 µL lysis buffer (10 mmol/L HEPES [N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid], 10 mmol/L KCl, 1.5 mmol/L MgCl2, and 0.5 mmol/L dithiothreitol [DTT], pH 7.9) with 0.2% Nonidet P-40 (NP-40) and protease inhibitor cocktail for 1 min on ice. After being microcentrifuged for 1 min at 2500×g, the supernatants were collected as cytoplasmic protein extracts. The pellets were washed with lysis buffer without NP-40, resuspended in 150 µL extraction buffer (20 mmol/L HEPES, pH 7.9, 420 mmol/L NaCl, 0.5 mmol/L DTT, 0.2 mmol/L EDTA, and 25% glycerol), and incubated for 20 min on ice. After centrifuged at 12 000×g for 10 min, the supernatants were collected as nuclear protein extracts.
RNA preparation and semiquantitative RT–PCR Semiquantitative RT–PCR with GAPDH as an internal control was performed to examine the expression of messenger RNA for the PI3Kp110α (PIK3CA). Total RNA (2 µg) from MUTZ-1 cells was reverse transcribed into single-stranded cDNA in a 20 µL reaction mixture containing 10 mmol/L dNTP, 1 µg oligo (dT) primer, and 200 IU M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA). The mixture was incubated at 42 ºC for 1 h, and then reverse transcriptase was inactivated by heating the reaction mixture to 95 ºC for 10 min. PCR amplification was carried out with 2 µL cDNA product in a 25 µL reaction volume containing 3 pmol of each specific oligonucleotide primer, 10 mmol/L dNTP, and 1.5 IU Taq DNA polymerase. For all of the reactions, preliminary experiments were performed to determine the number of PCR cycles that preceded saturation. The sequences of the primers, product size, and optimized number of PCR cycles for the PIK3CA and GAPDH expression analyses were: (1) PIK3CA, forward primer: 5’-TGTGGGACTTATTGAGG-3’; reverse primer: 5’-CACCATGATGTGCATCATTCA-3’ (GenBank accession no: Z29090; product size: 617 bp). The thermal cycle unit was programmed for 35 cycles at 95 ºC for 1 min, 61 ºC for 1 min, then 72 ºC for 2 min; and (2) GAPDH, forward primer: 5’-GAAGGTGAAGGTCGGAGTC-3’; reverse primer: 5’-GAAGATGGTGATGGGATTTC-3’ (GenBank accession no: NM-002046; product size: 225 bp).The thermal cycle unit was programmed for 26 cycles at 94 ºC for 30 s, 57 ºC for 45 s, then 72 ºC for 55 s. PCR products were separated by electrophoresis on a 1.8% agarose gel and visualized after ethidium bromide staining over UV light.
Antitumoral effect of CDA-2 on MUTZ-1 cell xenografted serious combined immunodeficiency disease mice Tumors models were established by injection of MUTZ-1 cells (1×107 cells/animal subcutaneously into the subscapularis of the serious combined immunodeficiency disease [SCID] mice) to 4-week-old male SCID mice (National Rodent Laboratory Animal Resource, Shanghai Branch, China). Treatments were initiated when tumors reached a mean group size of approximately 100 mm3. Tumor volume (mm3) was measured with calipers and calculated as (W2×L)/2, where W is the width and L is the length. The tumor growth inhibition rate was calculated by using the following formula: Inhibition Rate (IR) (%)=(1–TWt/TWc)×100, where TWt and TWc are the mean tumor weight of treatment and control groups. SCID mice was administered intraperitoneally with CDA-2 (1 and 4 g/kg) dissolved in 0.9% NaCl and 0.9% NaCl once every day. The weight and tumor volume of the mice were recorded every 3 d until the animals were killed. Animal care was in accordance with institutional guidelines.
Statistical analysis All results were given as mean±SD. Statistical significance (P<0.05) was assessed by ANOVA followed by Student’s t-test.
Results
CDA-2 inhibited the proliferation of MUTZ-1 cells as well as human leukemia cell lines Using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, we determined the effect of CDA-2 on MUTZ-1 cells and human leukemia cell lines as well as PBMC from 3 normal volunteers. The IC50 (50% inhibitory concentration) was measured in these cells treated with CDA-2 at concentrations ranging from 2 to 16 g/L for 24–72 h. CDA-2 exerted substantial growth inhibition in MUTZ-1 cells and human leukemia cell lines. The 24 and 48 h IC50 values showed 4.82 and 2.92 g/L in MUTZ-1 cells and approximately 5.51 (24 h), and 3.79 g/L (48 h) in myeloid leukemia cells, including HL-60, Kasumi-1, KG-1, U937, NB4, and K562 cell lines. The sensitivity of malignant lymphoid cell lines to CDA-2 was lower than those of MUTZ-1 cells and myeloid leukemia cells, with 24 and 48 h IC50 at 6.59 and 4.74 g/L, respectively (Figure 1A).
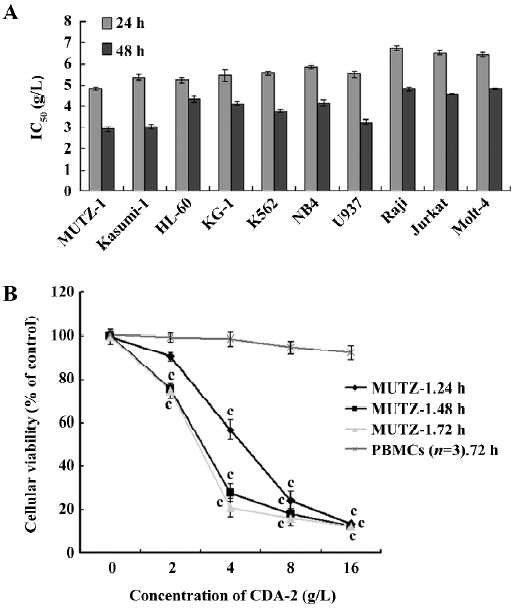
The MUTZ-1 cells were most sensitive to CDA-2, and cell growth was inhibited in a time- and dose-dependent manner, corresponding to the reduced cell viability (Figure 1B). In contrast, CDA-2 did not induce cytotoxicity in PBMC from the 3 normal volunteers (IC50=103.92 g/L, P<0.01; Figure 1B).
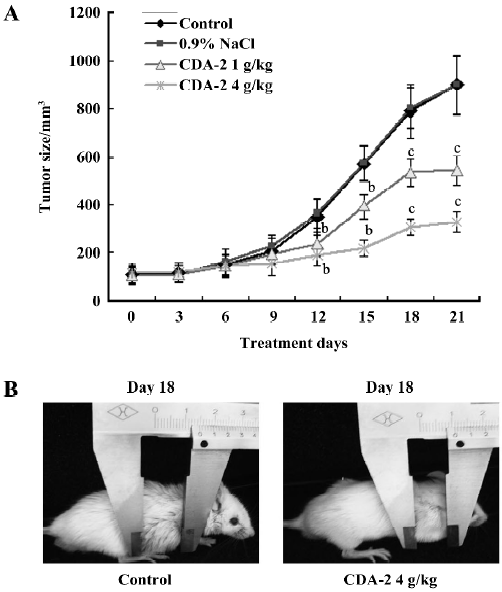
CDA-2 inhibited tumor growth in MUTZ-1 xenografted SCID mice The in vivo antiproliferative activity of CDA-2 was evaluated using human tumor models xenografted in SCID mice. CDA-2 inhibited tumor growth in a dose-dependent manner (Figure 2A). Compared with the control group, the tumor growth was significantly inhibited (Figure 2B; P<0.05-0.01) from d 12 to 21 in the groups treated with CDA-2 with tumor inhibition rates at 38.6% and 56.8% in the CDA-2 1 g/kg and 4 g/kg treatment groups, respectively. There was no significant body weight change in the SCID mice during the experiment.
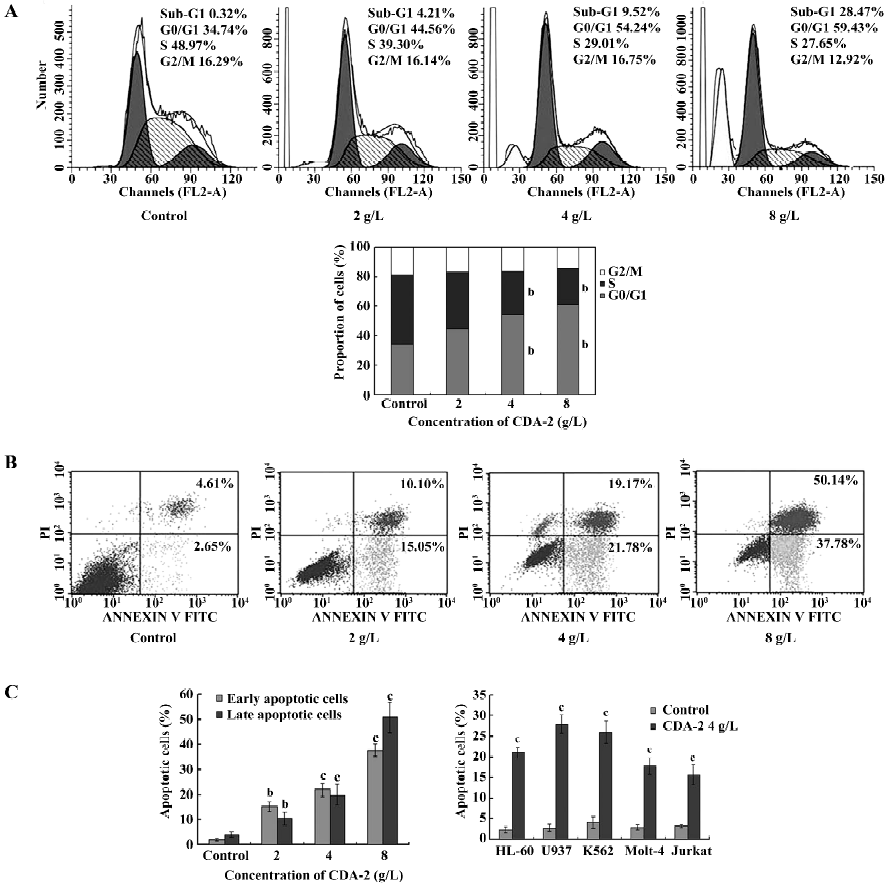
Inhibition of MUTZ-1 cell proliferation was due to G1 arrest and apoptosis induction To test whether there was an induction of cell growth arrest of CDA-2 treatment in MUTZ-1 cells, cell cycle analyses were performed. As depicted in Figure 3A, CDA-2 treatment resulted in an increased number of cells in the G1 phase of cell cycle, accompanied by a concomitant reduction of cells at the S phase of cell cycle in a dose-dependent manner. The apoptotic nature of cell death induced by CDA-2 was further confirmed by Annexin-V binding (Figure 3B). The percentage of early apoptotic and late apoptotic cells in the MUTZ-1 cell line were increased. These results indicated that CDA-2 induced G1 arrest and triggered apoptosis in MUTZ-1 cells. CDA-2-induced apoptosis was also verified in human leukemia cell lines, including HL-60, U937, K562, Molt-4, and Jurkat cells (Figure 3C).
Apoptosis triggered by CDA-2 was mediated via caspases-3, -8, and -9 and PARP cleavage, with caspase-3 a potential target of CDA-2-induced apoptosis To understand the mechanisms of CDA-2-induced cell death in MUTZ-1 cells, we examined the activation of caspases -3, -8, and -9, as well as the cleavage of PARP in MUTZ-1 cells. Treatment with CDA-2 induced marked activation of caspases-3 and -9 in a dose-dependent manner; however caspase-8 was activated only at CDA-2 concentrations greater than 4 g/L (Figure 4A). The cleavage of PARP, which is a substrate for the activated caspase-3, was also observed after CDA-2 treatment (Figure 4A).
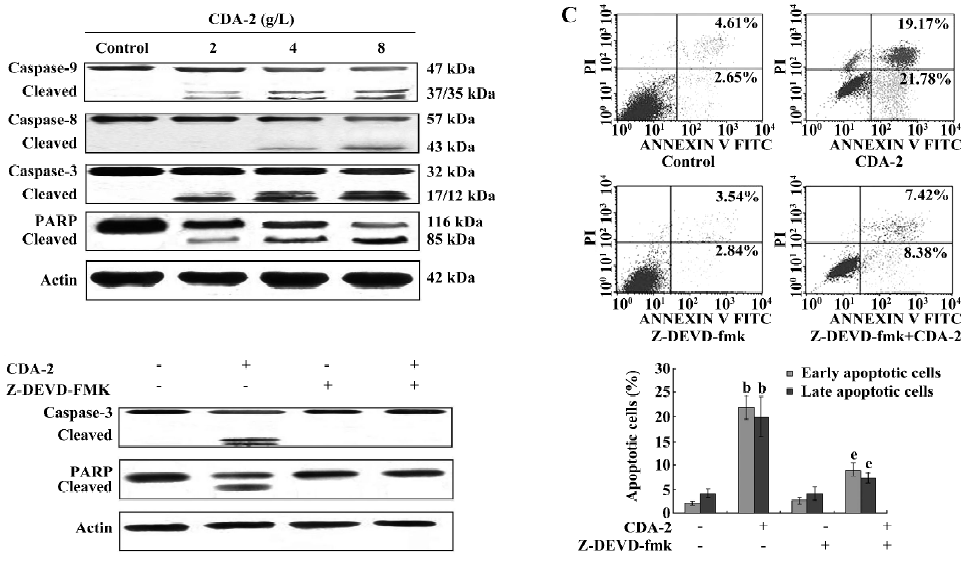
We next attempted to determine whether caspase-3 was likely to play an important role in CDA-2-induced apoptosis by treating cells with the specific caspase-3 inhibitor, Z-DEVD-fmk[12]. The inhibition of caspase-3 activity by pretreatment with 40 µmol/L Z-DEVD-fmk significantly decreased the apoptotic cells following CDA-2 treatment (Figure 4B). Furthermore, CDA-2 treatment was associated with the appearance of caspase-3 and PARP cleavage products; however, a significant decrease in these products was observed following pretreatment with Z-DEVD-fmk (Figure 4C). These results suggested that CDA-2-induced apoptosis might be caspase-3-dependent.
Bcl-2 family,the inhibitor of apoptosis(IAP) family, and FLICE-like inhibitory proteinlong (FLIPL) were involved in CDA-2-induced apoptosis The Bcl-2 family and the inhibitor of apoptosis (IAP) family members could ultimately inhibit or promote apoptosis. Therefore, we investigated whether CDA-2 treatment could modulate the expression of these pro- and anti-apoptotic proteins. After treatment with CDA-2 for 24 h, there was a dose-dependent decrease in the level of Bcl-2 and dose-dependent increase in the level of Bax protein (Figure 5A), which activated caspase-9 by decreasing the Bcl-2/Bax ratio (Figure 5B). Bad, another regulatory companion protein of Bcl-2[13], appeared to be unchanged after exposure to CDA-2, while the phosphorylation state of Bad was altered (Figure 5A,5C). Importantly, cleaved Bid was observed after CDA-2 dosage greater than 4 g/L (Figure 5A), which was in accordance with the activation of caspase-8, suggesting that cross-talk of apoptotic signaling from caspases-8 to -9 occurs[14]. Increased ratios of cells with depolarized mitochondrial membranes were also shown in these apoptotic cells, which indicated that mitochondria were involved in the CDA-2-induced apoptosis (Figure 5D). Finally, the release of the Smac protein was observed in response to this agent (Figure 5A).
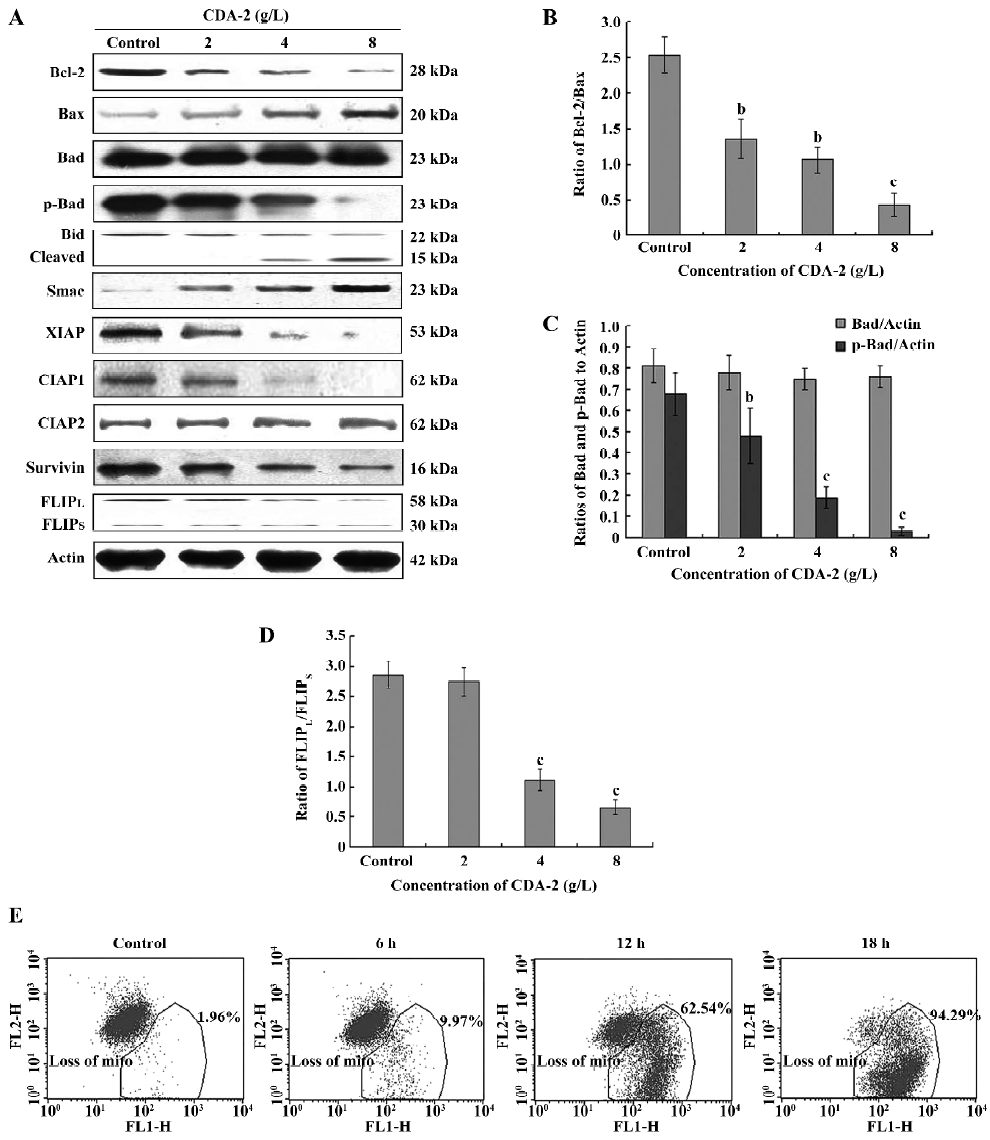
Because Smac reportedly promotes apoptosis by binding to and antagonizing the IAP family multiple proteins, including XIAP, CIAP, and survivin[15], the expression of these proteins was examined. The downregulation of XIAP, CIAP1, and survivin was observed in CDA-2-treated cells, whereas the expression of CIAP2 did not change (Figure 5A).
The FLIP protein was an antiapoptotic molecule which interfered with the receptor-mediated activation of caspase-8[16]. In MDS, the FLICE-like inhibitory proteinlong (FLIPL) and FLICE-like inhibitory proteinshort (FLIPS)splice variants was dysregulated. Therefore, we examined the expression of the FLIP protein in MUTZ-1 cells. MUTZ-1 cells had a high expression of FLIPL and a low expression of FLIPS, which was in accordance with the FLIP expression in high-risk MDS[17]. CDA-2 treatment could downregulate the expression of FLIPL when CDA-2 concentrations were greater than 4 g/L, which led to a decrease in the FLIPL/FLIPS, ratio and induced the activation of caspase-8.
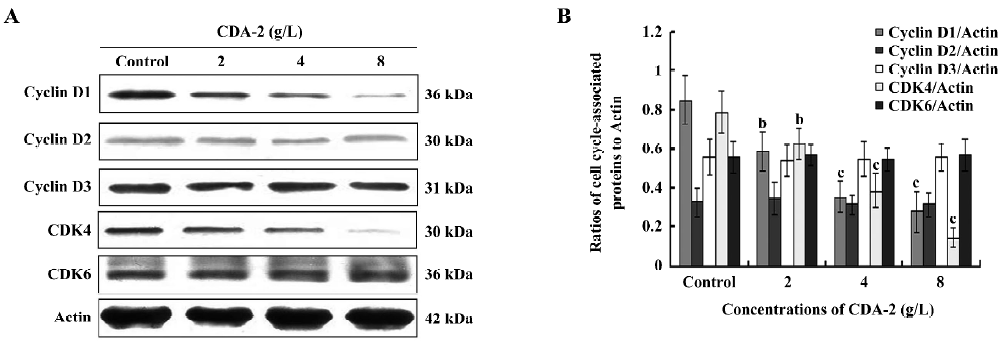
Cell cycle-associated proteins, cyclin D1 and CDK4, were involved in CDA-2-induced G1 arrest The levels of the cell cycle-associated proteins were determined by Western blotting. As shown in Figure 6, CDA-2 decreased the abundance of cyclin D1 and CDK4 in the protein levels in a dose-dependent manner, which was in accordance with CDA-2-induced MUTZ-1 cell line G1 arrest. However, the protein levels of cyclin D2, cyclin D3, and CDK6 were unchanged. Such data suggests that CDA-2 induced G1 arrest involved cyclin D1 and CDK4 cell proteins.
CDA-2 inactivates NF-κB by preventing NF-κB phosphorylation and nuclear translocation in MUTZ-1 cells Since the Bcl-2 family, IAP family, FLIPL, and cyclin D1 proteins are all known to be transcriptionally regulated by NF-κB[18-23], we next examined the NF-κB (P65) nuclear translocation in MUTZ-1 cells treated with 2-8 g/L CDA-2. The nuclear content of activated NF-κB (p-P65) was decreased with its accumulation in the cytoplasm in its inactive manner after CDA-2 treatment (Figure 7A).
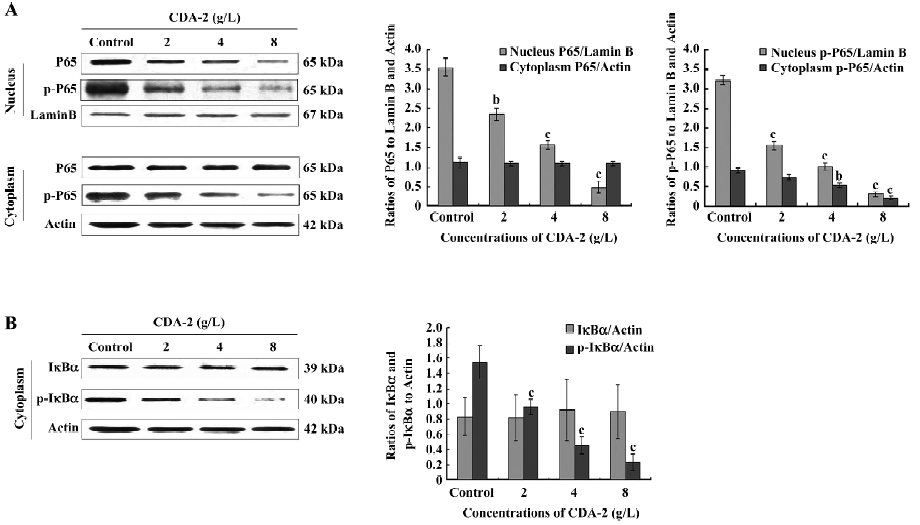
It has been shown that the nuclear translocation of NF-κB occurs as a result of NF-κB phosphorylation as well as IκBα phosphorylation and degradation. In our study, NF-κB and IκBα phosphorylation was inhibited, while total NF-κB and IκBα remained unchanged in the CDA-2-treated MUTZ-1 cell cytoplasm (Figure 7), which therefore inhibited NF-κB nuclear translocation.
Inactivation of the PI3K/Akt survival pathway by decreased PIK3CA transcription was associated with CDA-2-mediated apoptosis We further investigated the PI3K/Akt survival pathway, which had an effect on caspase-9, p-Bad, p-IκBα, and the IAP family. After treatment with CDA-2 for 24 h, there was a dose-dependent decrease in the level of PI3Kp110α, p-Akt, while total Akt remained unchanged (Figure 8A). We next examined the phosphorylation level of Akt in some myeloid leukemia cell lines, including K562, Kasumi-1, and KG-1, as well as some malignant lymphoid cell lines, including Jurkat, Molt-4, and Raji. The MUTZ-1, Kasumi-1, and KG-1 cell lines all had a high expression of p-Akt, with MUTZ-1 having the highest expression, which was in accordance with the sensitivity to CDA-2 (Figure 8B). The inhibition of PI3Kp110α expression was further confirmed at the mRNA level in a time- and dose-dependent manner after exposure to CDA-2 in the setting of induction of apoptosis in MUTZ-1 cells (Figure 8C).
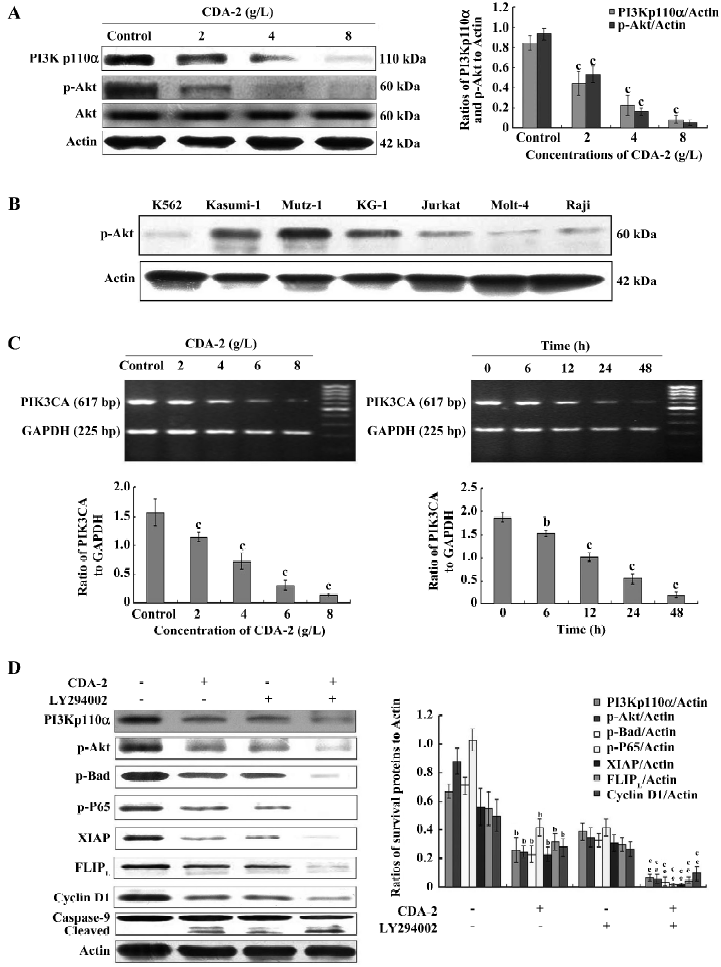
To further verify the role of the PI3K/Akt pathway on CDA-2-induced apoptosis, MUTZ-1 cells were treated with CDA-2 in the presence or absence of PI3K inhibitor LY294002 (30 µmol/L). CDA-2 combined with LY294002 significantly inhibited the expression of p-Akt, and therefore, downregulated p-Bad, p-P65, XIAP, FLIPL, cyclin D1, and activated caspase-9 (Figure 8D).
Discussion
Searching for novel therapeutics for treating high-risk MDS is not limited to traditional low dosage chemotherapies. Recently, the medicinal use of urine preparations in anticancer has been studied, and various mechanisms have been proposed to explain their effects. In our experiments, we first found that CDA-2, a urinary preparation, exerted potent antiproliferative activity in the MDS–RAEB-derived cell line, MUTZ-1. However, it exhibited no cytotoxicity of PBMC from normal volunteers. We further evaluated the effects of CDA-2 on MUTZ-1 xenografted SCID mice. The results of the in vivo experiments demonstrated the potent anticancer activity of CDA-2 in inhibiting tumor growth in a dose-dependent manner, without any significant influence on the body weight of mice. On the basis of the in vitro studies, it was most likely that the anticancer effect on MUTZ-1 xenografted SCID mice was due to CDA-2-mediated apoptosis, and the results of the apoptosis assay were consistent with our hypothesis.
Furthermore, we identified the mechanisms involved in the process of CDA-2-induced apoptosis and our work revealed the obvious changes of the expression levels of several survival proteins that play key roles in apoptosis. Apoptosis is confirmed by the activation of the caspase cascade, which is a crucial gateway involved in the execution of apoptosis in a variety of cellular systems. Caspase-3 is an ultimate executioner of the caspase family, which is essential for the nuclear changes associated with apoptosis, including chromatin condensation[24]. PARP is a highly conserved nuclear enzyme that binds tightly to DNA and plays a role in DNA repair, recombination, proliferation, and genomic stability[25,26]. It is also known that when the caspase cascade is activated, caspase-3 disassembles PARP into cleaved fragments. Therefore, the appearance of the cleaved PARP is solid proof of the activation of the caspase family. Our observation showed that cleaved caspase-3 and PARP appeared after incubation of 2-8 g/L CDA-2 for 24 h. The caspase-3 inhibitor Z-DEVD-fmk (40 µmol/L) could significantly block CDA-2-induced caspase-3 activation and PARP cleavage, which indicated the activation of caspase-3 was close related to CDA-2-induced apoptosis.
The activation of the PI3K/Akt survival pathway is one of the critical steps in cell survival through the suppression of apoptosis[27,28]. Recently, activated Akt kinase has been demonstrated in high-risk MDS patients[29]. The activated Akt inhibits apoptosis by multiple mechanisms, including phosphorylation of Bad, caspase-9, NF-κB, and the modulation of the IAP protein family[30]. Inactive cytosolic Akt is recruited to the plasma membrane and is activated by phosphorylation at serine-473 and threonine-308 in response to growth factors or cytokines[31,32]. The phosphorylation of Akt is mediated by the direct binding of lipid second messengers, such as phosphatidylinosital-3,4-biphosphate (Ptdins(3,4)P2) and phosphatidylinositol-3,4,triphosphate (Ptdins(3,4,5)P3) that are generated by PI3K[33]. The PIK3CA gene, located at 3q26, encodes the p110α catalytic subunit of PI3K. This gene has been proposed as a putative oncogene in solid tumors and its amplification gives rise to the activation of Akt[34]. Our preliminary data showed that the activation of Akt was highest in the MUTZ-1 cell line, which might be strongly associated with the sensitivity of CDA-2 in MUTZ-1 cells. Our data also showed that CDA-2 inhibited the activation of Akt through the inhibition of PIK3CA gene transcription. The blockade of Akt signaling by the PI3K inhibitor LY294002 (30 µmol/L) resulted in the inhibition of Akt phosphorylation in MUTZ-1 cells. When CDA-2 was combined with LY294002, the ability of CDA-2 to induce apoptosis of MUTZ-1 cells was potentiated, and the phosphorylation of Akt was significantly downregulated, which indicated that the inactivation of the PI3K/Akt survival pathway played an important role in CDA-2-induced MUTZ-1 cell death.
The PI3K/Akt pathway can transiently associate with the IκB kinase, which will then phosphorylate the inhibitory element IκB, thereby releasing NF-κB to translocate to the nuclear, while IκB is degraded via the proteasome pathway[35]. The constitutive activation of NF-κB is observed in high-risk MDS[36,37], and NF-κB regulates the expression of many genes important for apoptosis and the survival of cancer cells, including Bcl-2, the IAP family[38], as well as FLIPL[22]. In our studies, we found that CDA-2 could inhibit NF-κB activity through an impact on NF-κB phosphorylation and nuclear translocation. CDA-2 combined with LY294002 could significantly affect NF-κB phosphorylation. It was confirmed that CDA-2 had the ability to negatively regulate NF-κB activity through the inactivation of the PI3K/Akt survival pathway. Given the expression of NF-κB in MDS cells, but not normal primitive cells, CDA-2 can induce cancer-specific apoptosis through the inactivation of the PI3K/Akt/ NF-κB pathway.
The NF-κB pathway has been also implicated in regulating cell cycle progression, since NF-κB activity is induced as cell pass from the G0 phase into the G1 phase of the cell cycle[39]. Recently it has been shown that NF-κB transcriptionally regulates the expression of the cyclin D1 gene[40], and the cyclin D1 protein interacts with cyclin-dependent kinases, which phosphorylate and inactivate the retinoblastoma tumor suppressor[41]. In our studies, we found that CDA-2 could decrease the expression of cyclin D1 and CDK4 in a dose-dependent manner. CDA-2 combined with LY294002 could significantly inhibit the expression of cyclin D1. Thus it is possible that CDA-2 inhibits the progression of the cell cycle to the G1 phase by targeting the PI3K/Akt/NF-κB signaling pathway and prevent the expression of cyclin D1.
The primary cause of treatment failures in high-risk MDS is the emergence of both resistant disease and early relapse. Among the most frequent causes of these phenomena are the defects in the mitochondrial-mediated apoptosis pathway[42]. This pathway is regulated by the Bcl-2 family of anti-apoptotic (Bcl-2, Bcl-xl, and Mcl-1) and pro-apoptotic (Bax, Bad, and Bak) proteins. Our data demonstrated that CDA-2-induced apoptosis was most likely mediated by the dephosphorylation of Akt and subsequently Bad, thereby destabilizing the stoichiometric relationship between the Bcl-2 family members and their regulatory proteins. In our study, aside from Bad dephosphorylation, the expressions of Bax proteins were enhanced greatly by CDA-2 in a dose-dependent manner, while the amounts of Bcl-2 were downregulated in a strong and rapid manner. As Bax has the ability to heterodimerize with the Bcl-2 protein, the ratio of Bcl-2 to Bax appears as one of the important markers for the occurrence of apoptosis[43]. In fact, Bcl-2 and Bax, represent both sensitive indicators of clinical outcome and potential targets of novel pro-apoptotic molecules designed to circumvent chemoresistance[44]. A higher Bcl-2/Bax ratio confers a very poor prognosis with decreased rates of complete remission and overall survival[45–47]. The data in our study showed that the Bcl-2/Bax ratio decreased in a dose-dependent manner, which suggests that CDA-2 might represent a new therapeutic regiment, capable of inducing apoptosis at an early treatment state and overcoming multidrug resistance in vivo. Mitochondria, which may play a pivotal role in the interaction of Bcl-2 and Bax, are also important indications of apoptosis[48]. Therefore, the depolarized mitochondrial membranes were observed during CDA-2-induced apoptosis. A dose-dependent activation of the initiator caspase, caspase-9, was also observed after CDA-2 treatment. We concluded that the Bcl-2 family and mitochondria were involved in CDA-2-induced apoptosis, which was associated with the PI3K/Akt/Bad pathway.
The IAP family is a family of physiological substrates of Akt that are stabilized to inhibit programmed cell death. IAP have direct effects on caspases-9 and -3[49]. Our findings confirmed that a decrease of XIAP, CIAP1, and survivin was a common mechanism of CDA-2-induced cell death. CDA-2 combined with LY294002 could significantly downregulate XIAP, CIAP1, and survivin. These results also suggested that Akt might modulate these survival proteins for the survival of MUTZ-1 cells. Since IAP family were frequently overexpressed in high-risk MDS and overt leukemia transformed from MDS[50], the ability of CDA-2 to reduce the levels of the IAP family made it a powerful inducer of apoptosis and overcame multidrug resistance. By decreasing the IAP family, CDA-2 also can lower the apoptotic threshold and thereby enhance cell death induced by chemotherapeutic agents[48].
FLIP is another antiapoptotic molecule, which is regulated by NF-κB. It interferes with the receptor-mediated activation of caspase-8. The expression of FLIPL and FLIPS splice variants is dysregulated in MDS. Specifically, levels of FLIPL increased and levels of FLIPS decreased with progression of MDS from refractory anemia (RA) to RAEB (or more advanced disease), leading to an increase in the FLIPL/FLIPS ratio[51]. The FLIPL/FLIPS ratio directly correlated with the levels of NF-κB activity[17]. Our findings confirmed that MUTZ-1 cells had a high expression of FLIPL and a low expression of FLIPS. CDA-2 treatment could induce a dose-dependent decrease in FLIPL when CDA-2 concentrations were greater than 4 g/L. CDA-2 combined with LY294002 could significantly decrease the expression of FLIPL, which suggested that the PI3K/Akt/NF-κB signaling pathway were involved in the CDA-2-induced inhibition of FLIPL.
In summary, this study provides evidence that CDA-2 induces significant cytotoxicity in the MDS–RAEB-derived cell line MUTZ-1 in vitro. The mechanisms of CDA-2 cytotoxicity are related to the induction of apoptosis, which appears to be regulated by the PI3K/Akt survival pathway involving the prevention of NF-κB nuclear translocation, downregulation of the IAP family, FLIPL, and the dephosphorylation of the Bad protein, triggering the activation of the caspase cascades. Moreover, the in vivo antitumoral activity of CDA-2 is demonstrated in the MUTZ-1 xenografted model. The potent effects of CDA-2 in MDS–RAEB-derived cells reported in this study warrant further investigations for the treatment of high-risk MDS.
Acknowledgements
We thank for Ever Life Pharmaceutical (Hefei, Anhui, China) who kindly supplied the CDA-2.
References
- Mufti G, List AF, Gore SD, Ho AY. Myelodysplastic syndrome. Hematology Am Soc Hematol Educ Program 2003.176-99.
- Look AT. Molecular pathogenesis of MDS. Hematology Am Soc Hematol Educ Program 2005.156-60.
- Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. World Health Organization classification of tumors. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon (France): IARC Press; 2001.
- Parker JE, Mufti GJ. The myelodysplastic syndromes: a matter of life or death. Acta Haematol 2004;111:78-99.
- Burzynski SR. Antineoplastons: history of the research (I). Drugs Exp Clin Res 1986;12 suppl1:1-9.
- Badria F, Mabed M, Khafagy W, Abou-Zeid L. Potential utility of antineoplaston A-10 levels in breast cancer. Cancer Lett 2000;155:67-70.
- Lin WC, Wu YW, Lai TY, Liau MC. Effect of CDA-II, urinary preparation, on lipofuscin, lipid peroxidation and antioxidant systems in young and middle-aged rat brain. Am J Chin Med 2001;29:91-9.
- Lin WC, Liao YC, Liau MC, Lii CK, Sheen LY. Inhibitory effect of CDA-II, a urinary preparation, on aflatoxin B(1)-induced oxidative stress and DNA damage in primary cultured rat hepatocytes. Food Chem Toxicol 2006;44:546-51.
- Yao CJ, Lai GM, Chan CF, Yang YY, Liu FC, Chuang SE. Differentiation of pheochromocytoma PC12 cells induced by human urine extract and the involvement of the extracellular signal-regulated kinase signaling pathway. J Altern Complement Med 2005;11:903-8.
- Steube KG, Gignac SM, Hu ZB, Teepe D, Harms D, Kabisch H, et al. In vitro culture studies of childhood myelodysplastic syndrome: establishment of the cell line MUTZ-1. Leuk Lymphoma 1997;25:345-63.
- Drexler HG. Malignant hematopoietic cell lines: in vitro models for the study of myelodysplastic syndromes. Leuk Res 2000;24:109-15.
- Fischer U, Schulze-Osthoff K. Apoptosis-based therapies and drug targets. Cell Death Differ 2005;12 Suppl 1:942-61.
- Urbano A, Gorgun G, Foss F. Mechanisms of apoptosis by the tyrphostin AG957 in hematopoietic cells. Biochem Pharmacol 2002;63:689-92.
- Chandrasekar B, Vemula K, Surabhi RM, Li-Weber M, Owen-Schaub LB, Jensen LE, . Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem 2004; 279: 20 221−33.
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000;102:43-53.
- Kerbauy DB, Deeg HJ. Apoptosis and antiapoptotic mechanisms in the progression of myelodysplastic syndrome. Exp Hematol 2007;35:1739-46.
- Kerbauy DM, Lesnikov V, Abbasi N, Seal S, Scott B, Deeg HJ. NF-kappaB and FLIP in arsenic trioxide (ATO)-induced apoptosis in myelodysplastic syndromes (MDSs). Blood 2005;106:3917-25.
- Sun Y. E3 ubiquitin ligases as cancer targets and biomarkers. Neoplasia 2006;8:645-54.
- Lee R, Collins T. Nuclear factor-kappaB and cell survival: IAPs call for support. Circ Res 2001;88:262-4.
- Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med 1998;188:211-6.
- Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene 2001;20:7342-51.
- Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol 2001;21:5299-305.
- Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kappaB in development and progression of human cancer. Virchows Arch 2005;446:475-82.
- Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kagi D, et al. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev 1998;12:806-19.
- Burkle A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. Bioessays 2001;23:795-806.
- Tong WM, Cortes U, Wang ZQ. Poly (ADP-ribose) polymerase: a guardian angel protecting the genome and suppressing tumorigenesis. Biochim Biophys Acta 2001;1552:27-37.
- Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004;18:189-218.
- Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol 2005;17:150-7.
- Nyàkern M, Tazzari PL, Finelli C, Bosi C, Follo MY, Grafone T, et al. Frequent elevation of Akt kinase phosphorylation in blood marrow and peripheral blood mononuclear cells from high-risk myelodysplastic syndrome patients. Leukemia 2006;20:230-8.
- Lee S, Choi EJ, Jin C, Kim DH. Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol Oncol 2005;97:26-34.
- Hayakawa J, Ohmichi M, Kurachi H, Kanda Y, Hisamoto K, Nishio Y, et al. Inhibition of Bad phosphorylation either at serine-112 via extracellular signal-regulated protein kinase cascade or at serine-136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res 2000;60:5988-94.
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev 1999;13:2905-27.
- Kennedy SG, Wagner AJ, Conzen SD, Jordán J, Bellacosa A, Tsichlis PN, et al. The PI3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev 1997;11:701-13.
- Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet 1999;21:99-102.
- Béraud C, Henzel WJ, Baeuerle PA. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-kappaB activation. Proc Natl Acad Sci USA 1999;96:429-34.
- Braun T, Carvalho G, Coquelle A, Vozenin MC, Lepelley P, Hirsch F, et al. NF-kappaB constitutes a potential therapeutic target in high-risk myelodysplastic syndrome. Blood 2006;107:1156-65.
- Sanz C, Richard C, Prosper F, Fernandez-Luna JL. Nuclear factor kappa B is activated in myelodysplastic bone marrow cells. Haematologica 2002;87:1005-6.
- Takada Y, Kobayashi Y, Aggarwal BB. Evodiamine abolishes constitutive and inducible NF-kappaB activation by inhibiting IkappaBalpha kinase activation, thereby suppressing NF-kappaB-regulated antiapoptotic and metastatic gene expression, up-regulating apoptosis, and inhibiting invasion. J Biol Chem 2005; 280: 17 203−12.
- Baldwin AS Jr, Azizkhan JC, Jensen DE, Beg AA, Coodly LR. Induction of NF-kappa B DNA-binding activity during the G0-to-G1 transition in mouse fibroblasts. Mol Cell Biol 1991;11:4943-51.
- Joyce D, Bouzahzah B, Fu M, Albanese C, D’Amico M, Steer J, . Integration of Rac-dependent regulation of cyclin D1 transcription through a nuclear factor-kappaB-dependent pathway. J Biol Chem 1999; 274: 25 245−9.
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995;81:323-30.
- Armstrong JS. Mitochondrial medicine: pharmacological targeting of mitochondria in disease. Br J Pharmacol 2007;151:1154-65.
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993;74:609-19.
- Del Poeta G, Venditti A, Del Principe MI, Maurillo L, Buccisano F, Tamburini A, et al. Amount of spontaneous apoptosis detected by Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML). Blood 2003;101:2125-31.
- Dlugosz PJ, Billen LP, Annis MG, Zhu W, Zhang Z, Lin J, et al. Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J 2006;25:2287-96.
- Sedletska Y, Giraud-Panis MJ, Malinge JM. Cisplatin is a DNA-damaging antitumour compound triggering multifactorial biochemical responses in cancer cells: importance of apoptotic pathways. Curr Med Chem Anticancer Agents 2005;5:251-65.
- Ryningen A, Ersvaer E, Oyan AM, Kalland KH, Vintermyr OK, Gjertsen BT, et al. Stress-induced in vitro apoptosis of native human acute myelogenous leukemia (AML)cells shows a wide variation between patients and is associated with low BCL-2:Bax ratio and low levels of heat shock protein 70 and 90. Leuk Res 2006;30:1531-40.
- Carter BZ, Mak DH, Schober WD, McQueen T, Harris D, Estrov Z, et al. Triptolide induces caspase-dependent cell death mediated via the mitochondrial pathway in leukemic cells. Blood 2006;108:630-7.
- Dan HC, Sun M, Kaneko S, Feldman RI, Nicosia SV, Wang HG, et al. Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP). J Biol Chem 2004;279:5405-12.
- Yamamoto K, Abe S, Nakagawa Y, Suzuki K, Hasegawa M, Inoue M, et al. Expression of IAP family proteins in myelodysplastic syndromes transforming to overt leukemia. Leuk Res 2004;28:1203-11.
- Benesch M, Platzbecker U, Ward J, Deeg HJ, Leisenring W. Expression of FLIP(Long) and FLIP(Short) in bone marrow mononuclear and CD34+ cells in patients with myelodysplastic syndrome: correlation with apoptosis. Leukemia 2003;17:2460-6.