
Effect of atorvastatin on non-ischemic heart failure and matrix metalloproteinase-2 and 9 in rats1
Introduction
3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, or statins, are widely used clinically as a lipid lowering agent. Many studies have demonstrated that statins prevent the development of heart failure in ischemic heart disease or after myocardial infarction[1–3], but those effects of statins are always considered beneficial because of its role on atherosclerotic plaque, such as decreasing plasma cholesterol levels, inhibiting platelet activation, stabilizing plaque, and preventing cell proliferation and migration. However whether statins also inhibit heart failure in non-ischemic heart disease and its mechanism for it is still controversial. Recently, some clinical and basic studies indicated that statins improved cardiac function and survival in non-ischemic heart failure[4–8]. Concerns have also been raised about the potential adverse effects of statins on heart failure, such as decreased serum cholesterol, which causes the worst outcomes in heart failure[9] and reduced ubiquinone (coenzyme Q-10)[10–12], which may adversely affect mitochondrial and cardiac muscle function.
Matrix metalloproteinases (MMP) are a family of zinc-dependent enzymes that play an important role in the degradation of extracellular matrixes. Changes in expression and activity of several MMP have been identified in failing myocardium, and pharmacological inhibition of some MMP have resulted in the suppression of heart failure[13–17]. However, MMP species are not uniformly upregulated in heart failure; it is dependent on different heart failure etiologies and different stimulus. For example, MMP-2 and 3 were only upregulated in human non-ischemic dilated cardiomyopathy (DCM); MMP-9 was upregulated while MMP-1 was downregulated in both non-ischemic and ischemic DCM[18].
Some animal strains and heart failure models have been used to demonstrate the mechanisms of statins on heart failure, such as spontaneously hypertensive rats, Dahl salt-sensitive rats[19], the aortic stenosis model, and the rapid-pacing-induced model. However, all of the models have their shortcomings and some do not closely mimic the alterations observed clinically in most patients. In the present study, we planned to use the arteriovenous (AV) fistula-induced heart failure model, a well-established and thoroughly characterized model of volume-overload in rats, to evaluate the effect of atorvastatin on heart failure and its role on myocardial MMP-2 and 9 expression and activity.
Materials and methods
Surgical preparation and experimental protocol This study was approved by our institutional animal research committee (Zhejiang University, Hangzhou, China) and conformed to the Guide for the Care and Use of Laboratory Animals published by the United States National Institute of Health (NIH publication, N
Echo study The rats were anesthetized with ketamine HCl (50 mg/kg), and transthoracic echocardiography was conducted at 18 weeks. All rats with the HP Sonos 100 (Hewlett-Packard Co, Stockton, CA, USA) with a 10 MHz imaging linear scan probe transducer. The heart was imaged at the level of the papillary muscles to obtain left ventricle (LV) wall thickness and fractional shortening. Three beats were averaged for each measurement.
We determined the LV end diastolic diameter (LVEDd) as the widest and the end systolic diameter (LVEDs) as the narrowest dimension in the M-mode recordings. LV fractioning shortening (FS) was calculated according to the following formula:
LV FS (%)=(LVEDd–LVEDs)/LVEDd×100
Zymography of MMP activity LV myocardial samples were homogenized in 2 mL of an ice-cold extraction buffer containing cacodylic acid (10 mmol/L), NaCl (0.15 mol/L), ZnCl (20 mmol/L), NaN3 (1.5 mmol/L), and 0.01% Triton X-100 (pH 5.0). The homogenate was then centrifuged (4 °C, 10 min, 12 000×g) and the supernatant was saved and stored at -80 °C.
The myocardial extracts were loaded onto SDS-PAGE gel containing 1 mg/mL gelatin. The gelatin was stirred and heated at 50 °C for 1 h before adding to the gel. The myocardial extracts, at a final protein content of 4 µg, were loaded onto the gels using a 3:1 sample buffer (10% SDS, 4% sucrose, 0.25 mol/L Tris-HCl, and 0.1% bromophenol blue, pH 6.8). The gels were run at 20 mA/gel, maintaining a running buffer temperature of 4 °C. After that, the gels were washed twice with 2.5% Triton X-100 for 30 min each on ice and incubated with substrate buffer (50 mmol/L Tris-Cl, 5 mmol/L CaCl2, 0.02% NaN3, and 1% Triton X-100, pH 8) at 37 °C for 18 h. After incubation, the gels were stained with 0.05% Brilliant Blue R-250 (Sigma, St Louis, MO, USA), destained with 10% acetic acid and 20% methanol (v/v), and digitized. The gelatinolytic activity was tested as specific MMP activity by adding EDTA and phenylmethylsulphonyl fluoride PMSF. EDTA inhibited gelatinolytic activity, but PMSF did not.
Immunoblot analysis Proteins were isolated from the LV tissue, and 50 µg of total proteins were analyzed on 10% SDS-PAGE under the reducing condition. The proteins were blotted onto Polyvinylidene fluoride PVDF membranes and incubated in 5% non-fat milk in PBS for at least 1 h. After incubation, the PVDF membranes were subjected to immuno-blot analysis with rabbit polyclonal antibodies to MMP-2 or MMP-9 (1:200 dilutions; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or actin (1:400 dilutions, Santa Cruz Biote-chnology, USA) Immune complexes were detected with appropriate horseradish peroxidase-conjugated secondary antibodies and quantitated with Quantity One Image software (Bio-Rad, Hercules, CA, USA).
RNA isolation and RT-PCR analysis Total RNA was extracted from the LV tissue using TRIzol reagent. Briefly, total RNA (2 µg) was converted to single stranded cDNA using a reverse transcription system (Promega, Madison, WI, USA). The target cDNA was amplified using the following sense primer and antisense primers for rat brain natriuretic peptide (BNP). Sense: 5'-GGA AAT GGC TCA GAG ACA GCT C-3'; antisense: 5'-AAG TCT CTC CTG GAT CCG GAA G-3'. For GAPDH, sense: 5'-AAG GTC GGA GTC AAC GGA TTT-3'; and antisense: 5'-AGA TGA TGA CCC TTT TGG CTC-3'. The amplification cycles were 95 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min. After 30 cycles, the PCR products were separated by electrophoresis on 1.8% agarose (106 bp for BNP and 352 bp for GADPH).
Morphological measurement The LV was fixed with 4% paraformaldehyde, embedded in paraffin, sectioned at a thickness of 6 μm, and stained with hematoxylin-eosin and van Gieson staining. The degree of collagen fiber accumulation was quantified blindly on 6 sections per animal (12 randomly selected fields per section), and the ratio of the van Gieson staining fibrosis area to the total myocardium area was calculated.
Statistical analysis Statistical analyses were performed with SPSS 10.0 software (SPSS Inc, Chicago, IL, USA). All grouped data were expressed as mean±SD. Grouped data comparisons were made by one-way ANOVA followed by Bonferroni post-hoc testing. A P value of <0.05 was considered statistically significant.
Results
Left ventricular dilation and hypertrophy The FIS rats developed remarkable left ventricular dilation and hypertrophy compared with the CON rats at 18 weeks. LVEDd, LVEDs, and heart weight HW/body weight (BW) increased significantly in the FIS rats compared with the CON rats. FS in the FIS rats (41.4%±4.5%) decreased significantly compared with the CON rats (55.1%±4.5%, P<0.01). Atorvastatin treatment suppressed left ventricular dilation and hypertrophy significantly. LVEDd, LVEDs, and HW/BW in the ATO rats decreased remarkably compared with the FIS rats. Atorvastatin treatment also improved FS from 41.4%±4.5% to 52.7%±4.2% (P<0.01; Table 1).
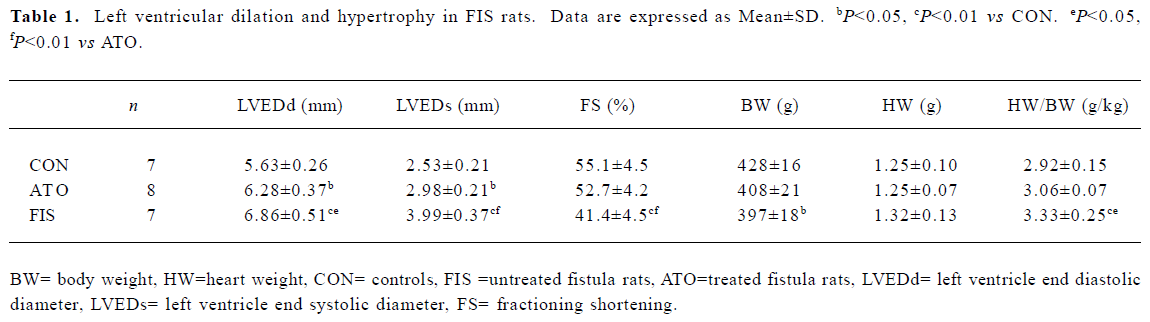
Full table
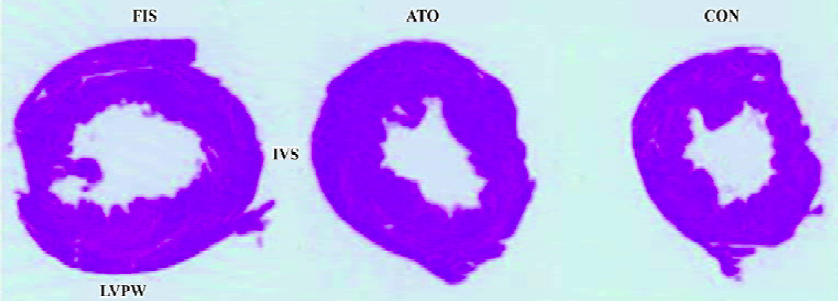
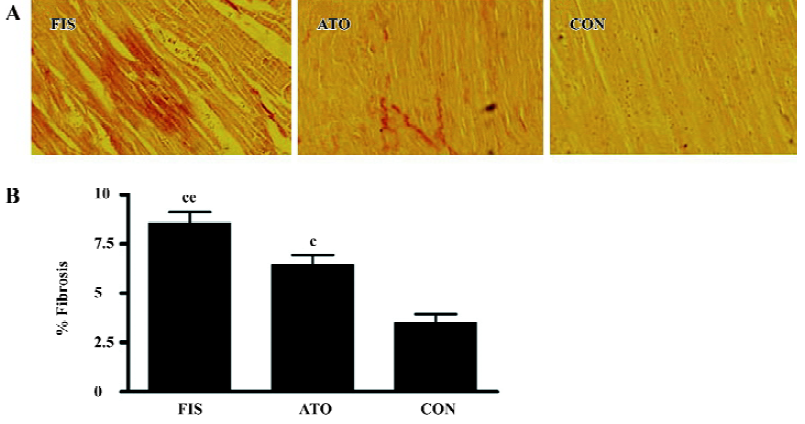
Marked left ventricular dilation and interstitial fibrosis were observed in the FIS group compared with the CON group. Atorvastatin treatment inhibited the left ventricular dilation and interstitial fibrosis significantly (Figures 1, 2). Atorvastatin reduced the ratio of fibrosis from 8.6%±1.4% in the FIS group to 6.4%±1.2% in the ATO group (P<0.05; Figure 2B).
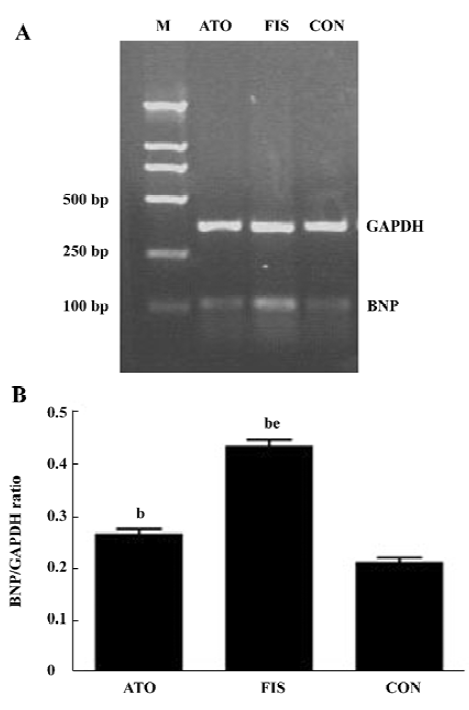
Left ventricular BNP mRNA level The mRNA levels of BNP increased about 2-fold in the FIS group compared with the CON group. Atorvastatin treatment reduced the mRNA levels of BNP significantly, and the ratio of BNP/GAPDH in the FIS group decreased from 0.43±0.03 to 0.27±0.03 in the ATO group (P<0.05, Figure 3A, 3B).
Effect of atorvastatin on myocardial MMP-9 and MMP-2 protein level and activity In the FIS rats, the MMP-9 protein level at 18 weeks after surgery was significant higher compared with the CON rats. Although the protein level of MMP-9 in the ATO rats was still significantly higher than the CON group, treatment with atorvastatin 17 week after surgery markedly inhibited the MMP-9 protein expression compared with the FIS rats (P<0.01, Figure 4A, 4B). Ator-vastatin treatment slightly, but still significantly, decreased the protein level of MMP-2 in the ATO rats compared with the FIS rats (P<0.05; Figure 4A, 4B). There were no significantly differences in the MMP-2 protein level between the ATO rats and the CON rats. Changes of MMP-9 and MMP-2 activity measured by zymography were consistent with the changes of MMP-9 and MMP-2 protein levels (Figure 5).
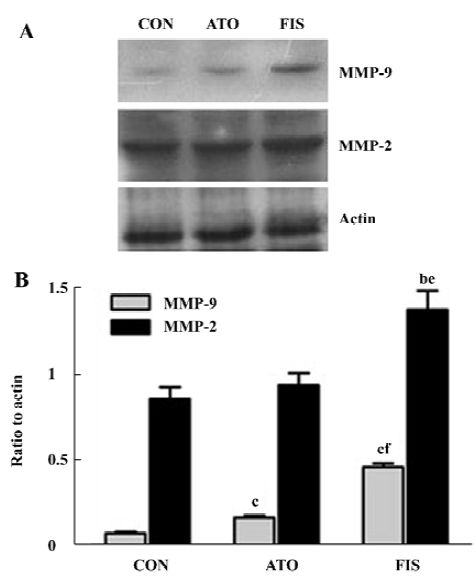
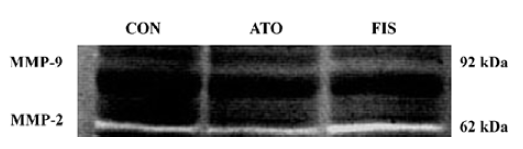
Discussion
The rats with an AV fistula can develop heart failure with normal sodium balance. This heart failure experimental model is characterized by the hemodynamic and neurohormonal changes, which closely mimic the alterations observed clinically in patients with heart failure[21,22]. Previous reports indicated that cardiac hypertrophy occurred within 1 week after AV fistula operation, while decompensate heart failure developed 8–16 weeks after the creation of AV fistula, characterized by circulatory congestion and decreased cardiac function[23]. The present study demonstrates that long-term administration of atorvastatin could prevent volume-overload-induced heart failure and left ventricular hypertrophy. More and more research has indicated that statins have therapeutic properties that are of potential benefit to heart failure with non-ischemic etiologies, irrespective of lipid levels. Rainer et al[24] demonstrated that simvastatin normalized autonomic neural control and reduced plasma norepinephrine in rapid-pacing-induced heart failure rabbits. Chen et al[20] demonstrated that simvastatin, initiated after hypertrophy, inhibited oxidative stress and prevented heart failure in rats with aortic stenosis. Inflammatory cytokines may also be involved in the role of statins on heart failure. Inflammatory cytokines, which are produced by activated macrophages, vascular wall cells, and cardiac myocytes, were elevated in heart failure. These cytokines, especially TNF-α and inter-leukin-6 (IL-6), exert negative inotropic effects and induce apoptosis in cardiac myocytes. Short-term simvastatin therapy has been tested to improve cardiac function and symptoms in patients with idiopathic dilated cardiomyopathy by reducing plasma concentrations of TNF-α and IL-6[4]. Failing myocardium of patients with DCM was characterized by upregulation of NADPH oxidase-mediated ROS release associated with increased Rac1 activity. Oral atorvastatin or pravastatin treatment could inhibit myocardial Rac1-GTPase activity[5]. These data suggest that antioxidative effects of statins may also be beneficial for heart failure. Other mechanisms of statins, such as promoting angiogenesis[25], increasing endothelial NO production, improving endothelial function and endothelial progenitor cell mobilization[3], have been demonstrated to take effect in heart failure after myocardial infarction, but whether these effects are also beneficial to heart failure with non-ischemia etiologies is still unknown.
Interstitial collagenases (such as MMP-1 and MMP-13), stromelysins (such as MMP-3), and gelatinases (such as MMP-2 and MMP-9) are expressed in mammalian myo-cardium. The relationship between MMP and the LV remodeling process has been demonstrated through the use of animal models of developing chronic heart failure, transgenic models, and the use of pharmacological MMP inhibition studies. Clinical research has also shown that MMP are involved in the process of LV remodeling and heart failure. The Framingham Heart Study demonstrated that plasma MMP-9 levels were associated with increased LV diastolic dimensions and increased wall thickness in men[26]. Levels of plasma MMP-2 were tested for an association with neurohormonal activation and levels of noradrenaline in human heart failure[27]. In ischemic heart failure, MMP-2 and MMP-13 levels increased, and statins were demonstrated to ameliorate ventricular remolding through inhibiting them[28]. Fibrosis during the progression of heart failure was also associated with increased MMP[29]. However, the types of MMP expressed in normal and congestive heart failure states remains unclear. The molecular basis for a selective portfolio of MMP to be increased within the failing human myocardium is likely due to the type, degree, and duration of the specific extracellular stimuli that are presented. Statins have been claimed to inhibit MMP in atherosclerosis[30–32], but its role on MMP in non-ischemic heart failure is not clear. This study found that atorvastatin prevented heart failure and LV hypertrophy, and these effects were associated with decreas-ed MMP-2 and MMP-9 protein expression and activity. Several possible mechanisms may be involved in the role of statins on the MMP system. Decreasing the secretion of inflammatory factor by statins is one of the possible mechanisms. For example, TNF-α, an important MMP regulator which activates MMP through the MAPK, NF-kappaB, and AP-1 pathways[33–35], may be involved in the role of statins on MMP. Turner NA et al[36] also demonstrated that simvastatin reduces MMP-9 secretion from human saphenous vein, smooth muscle cells by inhibiting isoprenoid formation and RhoA kinase. But whether it is the same in myocardium is unknown. More research needs to be done about the role of statins on different MMP species in heart failure.
In summery, our study demonstrated that atorvastatin may be beneficial for non-ischemic heart failure and may inhibit MMP-2 and 9 expression and activity.
References
- Bauersachs J, Galuppo P, Fraccarollo D, Christ M, Ertl G. Improvement of left ventricular remodeling and function by hydroxymethylglutaryl coenzyme a reductase inhibition with cerivastatin in rats with heart failure after myocardial infarction. Circulation 2001;104:982-5.
- Hognestad A, Dickstein K, Myhre E, Snapinn S, Kjekshus J. OPTIMAAL Investigators. Effect of combined statin and beta-blocker treatment on one-year morbidity and mortality after acute myocardial infarction associated with heart failure. Am J Cardiol 2004;93:603-6.
- Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A, Heineke A, et al. Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation 2004;110:1933-9.
- Node K, Fujita M, Kitakaze M, Hori M, Liao JK. Short-term statin therapy improves cardiac function and symptoms in patients with idiopathic dilated cardiomyopathy. Circulation 2003;108:839-43.
- Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M, et al. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation 2003;108:1567-74.
- Pliquett RU, Cornish KG, Peuler JD, Zucker IH. Simvastatin normalizes autonomic neural control in experimental heart failure. Circulation 2003;107:2493-8.
- Horwich TB, MacLellan WR, Fonarow GC. Statin therapy is associated with improved survival in ischemic and non-ischemic heart failure. J Am Coll Cardiol 2004;43:642-8.
- Mozaffarian D, Nye R, Levy WC. Statin therapy is associated with lower mortality among patients with severe heart failure. Am J Cardiol 2004;93:1124-9.
- Rauchhaus M, Coats AJ, Anker SD. The endotoxin-lipoprotein hypothesis. Lancet 2000;356:930-3.
- de Pinieux G, Chariot P, Ammi-Said M, Louarn F, Lejonc JL, Astier A, et al. Lipid-lowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 1996;42:333-7.
- Folkers K, Langsjoen P, Willis R, Richardson P, Xia LJ, Ye CQ, et al. Lovastatin decreases coenzyme Q levels in humans. Proc Natl Acad Sci USA 1990;87:8931-4.
- Strey CH, Young JM, Molyneux SL, George PM, Florkowski CM, Scott RS, et al. Endothelium-ameliorating effects of statin therapy and coenzyme Q10 reductions in chronic heart failure. Atherosclerosis 2005;179:201-6.
- Spinale FG, Coker ML, Thomas CV, Walker JD, Mukherjee R, Hebbar L. Time-dependent changes in matrix metalloproteinase activity and expression during the progression of congestive heart failure: relation to ventricular and myocyte function. Circ Res 1998;82:482-95.
- Coker ML, Thomas CV, Clair MJ, Hendrick JW, Krombach RS, Galis ZS, et al. Myocardial matrix metalloproteinase activity and abundance with congestive heart failure. Am J Physiol 1998;274:H1516-23.
- Nagatomo Y, Carabello BA, Coker ML, McDermott PJ, Nemoto S, Hamawaki M, et al. Differential effects of pressure or volume overload on myocardial MMP levels and inhibitory control. Am J Physiol 2000;278:H151-61.
- Peterson JT, Hallak H, Johnson L, Li H, O’Brien PM, Sliskovic DR, et al. Matrix metalloproteinase inhibition attenuates left ventricular remodeling and dysfunction in a rat model of progressive heart failure. Circulation 2001;103:2303-9.
- Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde M, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest 2000;106:55-62.
- Spinale FG, Coker ML, Heung LJ, Bond BR, Gunasinghe HR, Etoh T, et al. A matrix metalloproteinase induction/activation system exists in the human left ventricular myocardium and is upregulated in heart failure. Circulation 2000;102:1944-9.
- Ichihara S, Noda A, Nagata K, Obata K, Xu J, Ichihara G, et al. Pravastatin increases survival and suppresses an increase in myocardial matrix metalloproteinase activity in a rat model of heart failure. Cardiovasc Res 2006;69:726-35.
- Chen MS, Xu FP, Wang YZ, Zhang JP, Yi Q, Zhang HQ, et al. Statins initiated after hypertrophy inhibit oxidative stress and prevent heart failure in rats with aortic stenosis. J Mol Cell Cardiol 2004;37:889-96.
- Hatt PY, Rakusan K, Gastineau P, Laplace M. Morphometry and ultrastructure of heart hypertrophy induced by chronic volume overload (aorto-caval fistula in the rat). J Mol Cell Cardiol 1979;11:989-98.
- Pieruzzi F, Abassi ZA, Keiser HR. Expression of renin-angiotensin system components in the heart, kidneys and lungs of rats with experimental heart failure. Circulation 1995;92:3105-12.
- Wang X, Ren B, Lui S, Sentex E, Tappia PS, Dhalla NS. Characterization of cardiac hypertrophy and heart failure due to volume overload in the rat. J Appl Physiol 2003;94:752-63.
- Pliquett RU, Cornish KG, Peuler JD, Zucker IH. Simvastatin normalizes autonomic neural control in experimental heart failure. Circulation 2003;107:2493-8.
- Pourati I, Kimmelstiel C, Rand W, Karas RH. Statin use is associated with enhanced collateralization of severely diseased coronary arteries. Am Heart J 2003;146:876-81.
- Sundstrom J, Evans JC, Benjamin EJ, Levy D, Larson MG, Sawyer DB, et al. Relations of plasma matrix metalloproteinase-9 to clinical cardiovascular risk factors and echocardiographic left ventricular measures: the Framingham Heart Study. Circulation 2004;109:2850-6.
- Banfi C, Cavalca V, Veglia F, Brioschi M, Barcella S, Mussoni L, et al. Neurohormonal activation is associated with increased levels of plasma matrix metalloproteinase-2 in human heart failure. Eur Heart J 2005;26:481-8.
- Hayashidani S, Tsutsui H, Shiomi T, Suematsu N, Kinugawa S, Ide T, et al. Fluvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 2002;105:868-73.
- Boixel C, Fontaine V, Rucker-Martin C, Milliez P, Louedec L, Michel JB, et al. Fibrosis of the left atria during progression of heart failure is associated with increased matrix metallopro-teinases in the rat. J Am Coll Cardiol 2003;42:336-44.
- Fukumoto Y, Libby P, Rabkin E, Hill CC, Enomoto M, Hirouchi Y, et al. Statins alter smooth muscle cell accumulation and collagen content in established atheroma of watanabe heritable hyperlipidemic rabbits. Circulation 2001;103:993-9.
- Koh KK, Son JW, Ahn JY, Jin DK, Kim HS, Choi YM, et al. Comparative effects of diet and statin on NO bioactivity and matrix metalloproteinases in hypercholesterolemic patients with coronary artery disease. Arterioscler Thromb Vasc Biol 2002;22:e19-23.
- Furman C, Copin C, Kandoussi M, Davidson R, Moreau M, McTaqqiart F, et al. Rosuvastatin reduces MMP-7 secretion by human monocyte-derived macrophages: potential relevance to atherosclerotic plaque stability. Atherosclerosis 2004;174:93-8.
- Moon SK, Cha BY, Kim CH. ERK1/2 mediates TNF-alpha-induced matrix metalloproteinase-9 expression in human vascular smooth muscle cells via the regulation of NF-kappaB and AP-1: involvement of the ras dependent pathway. J Cell Physiol 2004;198:417-27.
- Wang BW, Chang H, Lin S, Kuan P, Shyu KG. Induction of matrix metalloproteinases-14 and -2 by cyclical mechanical stretch is mediated by tumor necrosis factor-alpha in cultured human umbilical vein endothelial cells. Cardiovasc Res 2003;59:460-9.
- Cho A, Graves J, Reidy MA. Mitogen-activated protein kinases mediate matrix metalloproteinase-9 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2000;20:2527-32.
- Turner NA, O’Regan DJ, Ball SG, Porter KE. Simvastatin inhibits MMP-9 secretion from human saphenous vein smooth muscle cells by inhibiting the RhoA/ROCK pathway and reducing MMP-9 mRNA levels. FASEB J 2005;19:804-6.