
Deferiprone protects the isolated atria from cardiotoxicity induced by doxorubicin
Introduction
Doxorubicin (DOX) is one of the anthracycline antibiotics widely used in cancer chemotherapy, yet it is limited by severe and cumulative cardiotoxicities despite its effectiveness[1,2]. The mortality of cardiomyopathy caused by DOX is as high as 40% in patients without treatment[3], and the incidences of congestive heart failure secondary to DOX-induced cardiomyopathy are 4%–36% in patients receiving cumulative dosages of 500–600 mg per square meter of body surface area[4]. The mechanism of DOX cardiotoxicity is still not well understood. It is widely accepted that oxidative stress and the production of free radicals are involved in DOX cardiotoxicity[4], in which iron was suggested to play an important role. DOX combines with Fe3+ in vivo. This DOX-Fe3+ complex can reduce oxygen to hydroxyl radical and other reactive oxygen species (ROS)[5], which initiate severe damage to cardiac tissues. Efforts have been made to prevent the DOX cardiotoxicity with a number of antioxidants and iron chelators such as melatonin[6] and ICL670A (deferasirox)[7]. So far there is only one agent, dexrazoxane (Zinecard), approved by the Food and Drug Administration (FDA) to prevent cardiomyopathy induced by DOX. However, dexrazoxane may potentiate the myelosuppression caused by DOX, and as a parenteral iron chelator, dexrazo-xane must be reconstituted with a sodium lactate injection and given slowly intravenously. Therefore, it is necessary and urgent to seek alternatives for clinical treatment besides dexrazoxane.
Deferiprone (L1, CP20) is the first oral iron chelator, licensed in the European Union for treating iron overload in thalassaemia patients unable to use deferoxamine[8]. Deferi-prone forms stable complexes with iron at a molar ratio of 3:1 (chelate:Fe3+)[9]. Several clinical trials have found that thalassaemia patients treated with deferiprone had higher ejection fractions than those treated with deferoxamine, and deferiprone was more effective than the latter in the removal of myocardial iron[10]. A previously published study of cultured hepatocytes showed that by scavenging superoxide radicals, both deferiprone and Fe3+-deferiprone complexes were able to protect the cells against hypoxia-reoxygenation injury effectively[11]. Further studies of cultured heart cells showed that deferiprone eliminated DOX-induced cardiac damage[12,13]. Together, these studies support the cardiac protective activity of deferiprone. The mechanism was possibly related to the iron chelating of deferiprone. As a small molecule, deferiprone is able to enter cardiac myocytes rapidly[13] and is capable of efficiently preventing free radical-based cardiotoxicity by quickly displacing iron from the complex with DOX[13]. Moreover, deferiprone and its complex with Fe3+ have antioxidant properties. It has been suggested that forming a redox complex with Fe3+ was the initial step for deferiprone to reduce oxidative damage: the complex could directly scavenge superoxide radicals with a free electron transference between them[11]. With the advantages of cheap synthesis and oral activity[14,15], deferiprone is promising to be an alternative besides dexrazoxane by improving compliance and easing financial burden.
The purpose of this study is to investigate the effects of deferiprone on DOX cardiotoxicity at tissue level with spontaneously-beating isolated paired atria from rats. Changes of atrial contractility (dF/dt) and mitochondrial structure induced by DOX were observed with or without the pretreatment of deferiprone. The experimental model we chose was due to 2 reasons. First, it was necessary to ensure that deferiprone could protect cardiac function from DOX before further studies in vivo since there are few reports about the effects of deferiprone on cardiac function. In addition, deferi-prone has a far shorter elimination time (elimination t1/2: 47 to 134 min[16]) than that of DOX (elimination t1/2: 20 to 48 h) in humans, which could interfere with the effects of deferiprone in vivo. Thus, we chose the model of isolated rat atria to avoid the possible interaction, which could also be useful for a rapid evaluation of DOX cardiotoxicity. Second, the effective dosages or dosage ratio of deferiprone to DOX is still unknown. As a chelator, deferiprone is administered at the dosage of 75 mg/kg per day in patients with high iron levels; this dosage might be too high or unsafe for DOX-treated patients with normal iron levels. In the present study, different concentrations of deferiprone were set in order to find the effective concentrations for later studies in vivo.
Materials and methods
Materials Deferiprone was purchased from Acros Organics (New Jersey, USA). DOX was purchased from Sigma Chemical Co (St Louis, Missouri, USA). The chemicals were dissolved in distilled water and were diluted in tissue bath solution to achieve the final concentrations.
Animals Forty eight adult male Sprague-Dawley rats at 10 weeks of age, weighing 350–450 g (Grade II), were obtained from the Department of Laboratory Animal Sciences of Fudan University (Shanghai, China). The rats were housed in a standard environment at a constant temperature of 22 °C under a 12-h light/dark cycle with free access to food and drinking water.
Preparation of spontaneously-beating isolated paired atria The rats were killed after anesthesia by urethane (1 g/kg) administered intraperitoneally. Both the left and right atria were dissected from the ventricles, cleaned of fatty tissues and mounted vertically at pH 7.4 at 30 °C in tissue bath filled with 6 mL Krebs-Henseleit buffer (in mmol/L: NaCl, 118; KCl, 4.7; KH2PO4, 1.2; MgSO4, 1.2; CaCl2, 1.25; NaHCO3, 25; glucose, 11) and aerated with 5% CO2 in oxygen. The paired atria were allowed to beat spontaneously under a resting tension of 1 g. An equilibrium period of 30 min was given before the application of drugs. Tension changes in the tissues were recorded every 10 min via a force-displacement transducer attached to SMUP-E Model Biosignal Processing System. Contractility (dF/dt) was assessed by recording the maximum rate of isometric force development. Control values (=100%) refer to the dF/dt before the addition of drugs[17].
Experimental protocols Spontaneously-beating isolated atria were divided randomly into 4 groups after the equilibrium period, and the experiment lasted 70 min.
DOX-treated atria were preincubated for 10 min without any drugs before incubation with DOX for 60 min. We tested the IC50 concentration of DOX (0.035 mmol/L) in a previous study[18], and then adjusted it to 0.03 mmol/L as the preliminary study showed a negative inotropic effect of 50% after 60 min. Deferiprone-pretreated atria were incubated with both deferiprone and DOX for 60 min; the addition of deferiprone was 10 min before that of DOX. The concentrations of deferiprone were 10 or 40 times higher than that of DOX (deferiprone:1.2 mmol/L+DOX:0.03 mmol/L or deferiprone:0.3 mmol/L+DOX:0.03 mmol/L). In a preliminary study, several concentrations of deferiprone had been tested with 0.03 mmol/L DOX. The results had not shown significant cardiac protection against DOX-induced damage on atria pretreated with 0.3 mmol/L deferiprone. Thus, 0.3 mmol/L was selected to be the low concentration of deferiprone. The control atria were incubated without any drugs during the whole process.
After the experiment, the cardiac tissues around the sinuatrial nodes were taken and fixed in 2.5% glutaraldehyde; the remains were homogenized (10%, w/v) at 4 °C in homogenization buffer consisting of 0.01 mol/L sucrose, 0.01 mol/L Tris-HCl, 0.1 mmol/L EDTA-2Na and 8% solution of NaCl, pH 7.4. Homogenates were distributed and stored at -70 °C until different assessments were conducted.
Histological studies Tissue samples were post-fixed with osmium tetroxide and embedded in araldite (epoxyresin). Semifine sections (0.5 µm) were stained with toluidine blue and observed by light microscopy; ultra-thin sections (80 nm) contrasted with uranyl acetate and lead citrate were examined by a Philips CM120 transmission electron microscope (Eindhoven, the Netherlands).
Determination of Cu, Zn-SOD and MDA levels Cu, Zn-SOD activity was measured with a spectrophotometer at 550 nm. Treatment with 2% sodium dodecyl sulfate (SDS) at 37 °C for 30 min eliminated Mn-SOD, but not Cu, Zn-SOD activity. After removing the excess sodium dodecyl sulfate by precipitation with 2% KCl, the supernate was assayed using the xanthine oxidase-cytochrome c method. One unit of SOD was defined as the amount of enzyme required to inhibit 50% of the rate of cytochrome c reduction at 25 °C, and data were expressed as units per microgramme[19].
MDA concentrations were determined after incubation at 95 °C and pH 3.4 with thiobarbituric acid as an indicator of lipid peroxidation. The pink colour produced by these reactions was measured at 532 nm in order to measure MDA concentrations[20].
Determination of SDH activity The activity of SDH was estimated by quantifying the deoxidization at the speed of 2, 6-dichlorophenol indophenol (DCPIP), which absorbs strongly at 600 nm when oxidized, but becomes colorless in its reduced form. The reaction mixture contained 0.12 mmol/L DCPIP, 10 mmol/L succinate and 20 µL of homogenate in a final volume of 2 mL (pH 7.8). The reaction was started by the addition of succinate and was monitored for 3 min. One activity unit of SDH was defined as 0.01 decreases in absorbance at 600 nm per mg protein per min[21].
Protein determination Protein determinations were performed by the Bradford method based on the specific binding of Coomassie blue G-250 dye to proteins. The binding caused a shift in the absorption maximum of the dye from 465 to 595 nm, and it was the increase in absorption at 595 nm that was monitored[22]. The Bradford reagent contained 0.01% w/v Coomassie Blue G-250. The protein concentrations were determined from a standard BSA curve prepared in parallel.
All of the colorimetry assays were performed with a UV-2401PC spectrophotometer (Shimadzu, Kyoto, Japan).
Statistical analysis All results were expressed as mean± SD and compared statistically by Student’s t-test. The numbers of animals (n) used per group are stated in Figures 1 and 2 and Table 1. The differences between means were considered significant if P<0.05.
Full table
Results
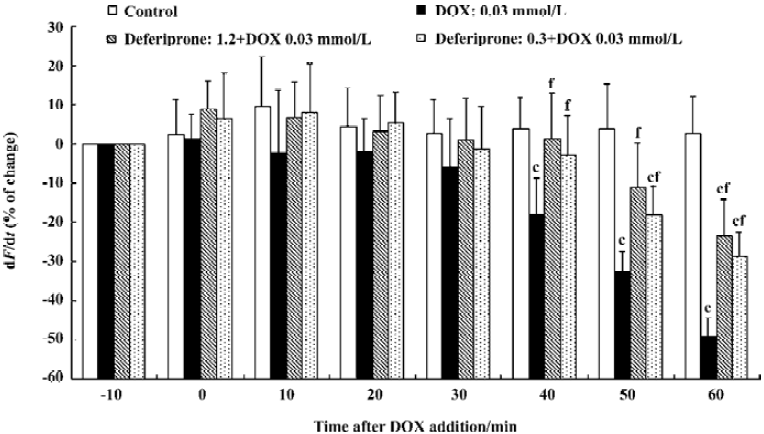
Effects of deferiprone on the contractility (dF/dt) of DOX-treated atria To determine whether deferiprone can protect cardiac function from DOX, we measured the contractility (dF/dt) of isolated atria with or without the presence of deferiprone (Figure 1). At the end of the equilibration period, the absolute value for dF/dt of the control atria was 19.3±5.3 g/s; DOX-treated atria was 21.6±6.1 g/s, and the deferiprone-pretreated atria (1.2 mmol/L or 0.3 mmol/L) was 20.4±5.9 g/s or 19.2±5.4 g/s. Incubation with 0.03 mmol/L DOX resulted in a gradual reduction of contractility of DOX-treated atria. Forty min after the addition of DOX, the contractility became significantly lower than that of the control atria (P<0.01), and decreased by 49.34%±4.79% (10.9±4.2 g/s) at the end of incubation. Neither the 1.2 mmol/L nor 0.3 mmol/L concentrations of deferiprone had significant effects on basal contractility 10 min after pretreatment, and the contractility of both deferiprone-pretreated atria did not decrease until 50 min after the addition of DOX. Furthermore, pretreatment of deferiprone did moderately attenuate DOX-induced negative inotropic effect. Compared with the DOX-treated atria, the contractility of the 1.2 mmol/L or 0.3 mmol/L concentrations of deferiprone-pretreated atria was significantly increased 40 min after the addition of DOX (P<0.01), and was 25.70% or 20.44% higher than that of DOX-treated atria at the end of the incubation, respectively (P<0.01).
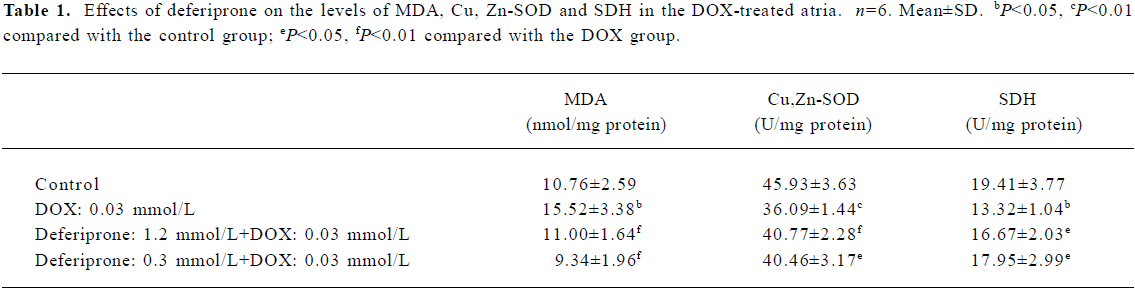
Effects of deferiprone on the level of MDA and Cu, Zn-SOD activity of DOX-treated atria Cu, Zn-SOD activity in the DOX-treated atria decreased by 21.42% (P<0.01) and MDA levels increased by 44.24% (P<0.05) compared with the control atria (Table 1). However, there was no statistically significant difference between the control and the deferiprone-pretreated atria. Moreover, Cu, Zn-SOD activity in the 1.2 mmol/L or 0.3 mmol/L deferiprone-pretreated atria were notably increased by 12.97% (P<0.01) or 12.11% (P<0.05) respectively. MDA levels were reduced by 29.12% (P<0.01) or 39.82% (P<0.01) compared with the DOX-treated atria.
Effects of deferiprone on SDH activity of DOX-treated atria The results (Table 1) show that SDH activity in the DOX-treated atria decreased by 31.38% compared with the control atria, (P<0.05). However, in the 1.2 mmol/L or 0.3 mmol/L deferiprone-pretreated atria, SDH activity increased by 25.15% or 34.76% (P<0.05) compared with the DOX-treated atria.
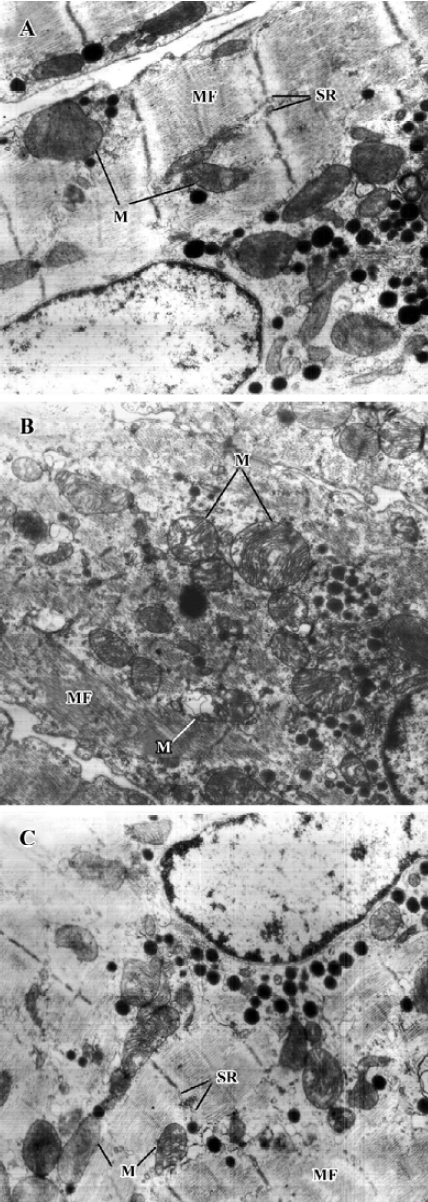
Effects of deferiprone on ultrastructure of DOX-treated atria Electron microscopic studies revealed that mitochondria swelling, disruption of mitochondrial crista and decreased electron density of the matrices were found in the cardiac myocytes of the DOX-treated atria. However, these lesions were ameliorated in the cardiac myocytes of the1.2 mmol/L deferiprone-pretreated atria (Figure 2).
Discussion
The present study differs from other published studies in that it provides evidence that deferiprone can protect against damage to cardiac contractility induced by DOX. The results of our study also suggest that deferiprone should be tested in vivo as a novel therapy for DOX-induced cardiotoxicity.
The effect of deferiprone on DOX cardiotoxicity was evaluated via spontaneously-beating isolated paired atria from rat. Recording changes of contractility of isolated atria can quantitate the effect of deferiprone on cardiac function. DOX was administered at the concentration of 0.03 mmol/L for 60 min in tissue bath solution. Atria contractility decreased gradually and nearly 50% of the contractility was lost 40 min after the addition of DOX, which was similar to the negative inotropic effect on the DOX-treated mouse atrium[18]. The contractility of both 1.2 mmol/L and 0.3 mmol/L deferiprone-pretreated atria decreased 50 min after the DOX addition, but they were 25.70% or 20.44% higher than that of the DOX-treated atria at the end of the incubation. The results indicate that pretreatment with deferiprone remarkably preserved the contractility of atria, although it could not entirely reverse the negative inotropic effects induced by DOX. A previously published study reported that deferiprone prevented cultured cardiac myocytes from DOX-induced damage monitored with the release of lactate dehydrogenase[13]. Our study further suggests that deferiprone can also protect cardiac contractile function from damage induced by DOX.
After incubation, we investigated the effects of deferi-prone on the mitochondria and SDH activity of the isolated atria treated with DOX. Cardiac myocytes contain the highest volume density of mitochondria in the body due to the extraordinary demand for continuous synthesis of ATP[23]. Unfortunately, mitochondria are also the target for DOX[24] and its complex with iron[12]. It was found that DOX had high affinity for cardiolipin, the phospholipid in the inner mitochondrial membrane[24]. This affinity results in the accumulation of DOX in cardiac myocytes with higher concentrations than that in other cells[25], and causes oxidative damage on mitochondrial membranes, the respiratory chain, and DNA[26–28]. On the other hand, the mitochondrial respiratory chain is the major source of superoxide, thus, mitochondria are more susceptible to oxidative damage than the rest of the cells[29]. SDH is a component of complex II of the respiratory chain and is the only tricarboxylic acid cycle enzyme embedded to the inner mitochondrial membrane that is essential for full respiratory activity. As a mitochondrial marker enzyme, the activity of SDH decreases when its vicinal thiols (SH group) are oxidized[30]. Previous studies have shown that the SH group is very sensitive and SDH is inactivated by oxidized DOX[31].
In our study, significant mitochondrial structural lesions and the decrease of SDH activity were observed in DOX-treated atria, which indicates the damage caused by DOX on the cardiac mitochondria. Conversely, the pretreatment of deferiprone resulted in fewer ultrastructure lesions and maintained SDH activity. These results suggest that deferiprone is able to protect both mitochondrial structure and function from DOX-induced damage and supports previous research[12] in which deferiprone had eliminated DOX-induced inhibition of mitochondrial function with a partial reduction of 14C-palmitate utilization in cardiac myocytes. However, other studies have reported that DOX has little effect on SDH activity[32,33]. The discrepancy may be attributed to the fact that in the current study, an acute DOX treatment lasting 60 min was performed in vitro, while in previous studies, DOX were repeatedly administered in vivo over several months or years. Nevertheless, these contradicting findings suggest that the inactivation of SDH might be an index of acute mitochondrial lesions rather than chronic ones.
We also investigated the antioxidant effects of deferiprone on the cardiac tissues of DOX-treated atria. Iron-based oxida-tive stress has been implicated to be responsible for DOX-induced cardiotoxicity[34]. Fe3+ reacts with DOX in a redox reaction after which the iron atom accepts an electron and a Fe2+ DOX-free radical complex is produced to stimulate ROS, including extremely damaging hydroxyl radicals[5,35]. In addition, the lack of natural antioxidants enhances the susceptibility of cardiac tissues to oxidative damage, which is suggested to be the inherent causes of DOX cardiotoxicity[36]. Assessments of SOD and the lipid peroxidation end product MDA have been always used as markers for oxidative damage. Cu, Zn-SOD is the major intracellular form of SOD, which is located in the cytoplasma and nucleus[37]. In this study, DOX induced a decrease in Cu, Zn-SOD activity and an increase in MDA levels in DOX-treated atria, which indicates that the oxidative damage caused by DOX are consistent with findings of previous studies[33,38]. Conversely, MDA levels were lower and Cu, Zn-SOD activity was maintained in the atria pretreated with deferiprone at concentrations 10 and 40 times higher than that of DOX. This result demonstrates the antioxidant property of deferiprone against DOX, and it is this antioxidant property that makes deferiprone surpass other iron chelators such as deferoxamine and ICL670A in cardio protection. Neither deferoxamine nor its complex with Fe3+ could scavenge superoxide radicals or reveal cytoprotective activity[11]. ICL670A was found to not protect cardiac myocytes against DOX[7]. The antioxidant property of deferiprone would be in part due to its direct superoxide radical scavenging[11] and iron-chelating activity[5]. It was demonstrated that deferiprone not only could quickly remove Fe3+ from its complex with DOX extracellularly, but also penetrated myocytes rapidly and displaced iron from its complex with the intracellularly-loaded fluorescent dye calcein[13]. Those findings suggest that like dexrazoxane, deferiprone is able to prevent the formation of Fe3+-DOX complex in the cardiac myocyte and as a result, prevent site-specific iron-based oxygen radical damage[13].
Both pretreatments of 1.2 mmol/L and 0.3 mmol/L deferiprone were shown to protect against DOX-induced cardiotoxicity without statistical differences between them. The results suggest that 10:1 is the approximate effective concentration rate of deferiprone to DOX in the current study, which must be further verified in vivo. However, in the preliminary study, pretreatment with 0.3 mmol/L deferiprone had not shown significant protection on the contractility after 60 min co-incubation with 0.03 mmol/L DOX. One of the possible reasons for this was that fewer rats were tested in preliminary study.
In conclusion, our findings indicate that deferiprone is able to improve cardiac contractility efficiently and protect mitochondrial structure and function from DOX-induced damage. The effectiveness of deferiprone could be due to defense capability against oxidative damage induced by DOX.
Acknowledgements
The authors thank Prof Jun PAN for her critical review and Mr Yin-xiang CAO and Mr Yuan QIAN for their excellent technical support on the software and adjustment of the biosignal processing system.
References
- Gianni L, Corden BJ, Myers CE. The biochemical basis of anthracycline toxicity and anti-tumor activity. Rev Biochem Toxicol 1983;5:1-82.
- Shan K, Lincoff AM, Young JB. Anthracycline-induced cardiotoxicity. Ann Intern Med 1996;125:47-58.
- Suter TM, Meier B. Detection of anthracycline-induced cardiotoxicity: is there light at the end of the tunnel? Ann Oncol 2002;13:647-9.
- Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. New Engl J Med 1998;339:900-5.
- Gianni L, Zweier JL, Levy A, Myers CE. Characterization of the cycle of iron-mediated electron-transfer from adriamycin to molecular-oxygen. J Biol Chem 1985;260:6820-6.
- Ahmed HH, Mannaa F, Elmegeed GA, Doss SH. Cardioprotective activity of melatonin and its novel synthesized derivatives on doxorubicin-induced cardiotoxicity. Bioorgan Med Chem 2005;13:1847-57.
- Hasinoff BB, Schroeder PE, Patel D. The metabolites of the cardioprotective drug dexrazoxane do not protect myocytes from doxorubicin-induced cytotoxicity. Mol Pharmacol 2003;64:670-8.
- Brittenham GM. Iron chelators and iron toxicity. Alcohol 2003;30:151-8.
- Kontoghiorghes GJ. Comparative efficacy and toxicity of desferrioxamine, deferiprone and other iron and aluminum chelating drugs. Toxicol Lett 1995;80:1-18.
- Anderson LJ, Wonke B, Prescott E, Holden S, Walker JM, Pennell DJ. Comparison of effects of oral deferiprone and subcutaneous desferrioxamine on myocardial iron concentrations and ventricular function in beta-thalassaemia. Lancet 2002;360:516-20.
- Moridani MY, O’Brien PJ. Iron complexes of deferiprone and dietary plant catechols as cytoprotective superoxide radical scavengers. Biochem Pharmacol 2001;62:1579-85.
- Link G, Tirosh R, Pinson A, Hershko C. Role of iron in the potentiation of anthracycline cardiotoxicity: Identification of heart cell mitochondria as a major site of iron-anthracycline interaction. J Lab Clin Med 1996;127:272-8.
- Barnabe N, Zastre JA, Venkataram S, Hasinoff BB. Deferiprone protects against doxorubicin-induced myocyte cytotoxicity. Free Radical Biol Med 2002;33:266-75.
- Kontoghiorghes GJ, Aldouri MA, Hoffbrand AV, Barr J, Wonke B, Kourouclaris T, et al. Effective chelation of iron in beta-thalassemia with the oral chelator 1,2-dimethyl-3-hydroxypyrid-4-one. Br Med J 1987;295:1509-12.
- Kontoghiorghes GJ, Neocleous K., Kolnagou A. Benefits and risks of deferiprone in iron overload in thalassaemia and other conditions: comparison of epidemiological and therapeutic aspects with deferoxamine. Drug Safety 2003;26:553-84.
- Kontoghiorghes GJ, Goddard JG, Bartlett AN, Sheppard L. Pharmacokinetic studies in humans with the oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one. Clin Pharmacol Ther 1990;48:255-61.
- Sterin-Borda L, Del Zar CF, Borda E. Differential CB1 and CB2 cannabinoid receptor-inotropic response of rat isolated atria: endogenous signal transduction pathways. Biochem Pharmacol 2005;69:1705-13.
- Voest EE, van Acker SABE, van der Vijgh WJF, van Asbeck BS, Bast A. Comparison of different iron chelators as protective agents against acute doxorubicin-induced cardiotoxicity. J Mol Cell Cardiol 1994;26:1179-85.
- Geller BL, Winge DR. A method for distinguishing Cu,Zn- and Mn-containing superoxide dismutases. Anal Biochem 1983;128:86-92.
- Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979;95:351-8.
- King T, Howard R. Preparation of succinate dehydrogenase and reconstitution of succinate oxidase. New York: Academic Press; 1967.
- Bradford MM. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal Biochem 1976;72:248-54.
- Goffart S, von Kleist-Retzow JC, Wiesner RJ. Regulation of mitochondrial proliferation in the heart: power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc Res 2004;64:198-207.
- Goormaghtigh E, Huart P, Praet M, Brasseur R, Ruysschaert JM. Structure of the adriamycin-cardiolipin complex. Role in mitochondrial toxicity. Biophys Chem 1990;35:247-57.
- Johnson BA, Cheang MS, Goldenberg GJ. Comparison of adriamycin uptake in chick embryo heart and liver cells an murine L5178Y lymphoblasts in vitro: role of drug uptake in cardiotoxicity. Cancer Res 1986;46:218-23.
- Demant EJF. Inactivation of cytochrome-c-oxidase activity in mitochondrial-membranes during redox cycling of doxorubicin. Biochem Pharmacol 1991;41:543-52.
- Palmeira CM, Serrano J, Kuehl DW, Wallace KB. Preferential oxidation of cardiac mitochondrial DNA following acute intoxication with doxorubicin. Biochim Biophys Acta 1997;1321:101-6.
- Lebrecht D, Setzer B, Rohrbach R, Walker UA. Mitochondrial DNA and its respiratory chain products are defective in doxorubicin nephrosis. Nephrol Dial Transpl 2004;19:329-36.
- Lenaz G, Cavazzoni M, Genova ML, D’Aurelio M, Pich MM, Pallotti F, et al. Oxidative stress, antioxidant defences and aging. Biofactors 1998;8:195-204.
- Lequoc K, Lequoc D, Gaudemer Y. Evidence for the existence of two classes of sulfhydryl groups essential for membrane-bound succinate dehydrogenase activity. Biochemistry 1981;20:1705-10.
- Muraoka S, Miura T. Inactivation of mitochondrial succinate dehydrogenase by adriamycin activated by horseradish peroxidase and hydrogen peroxide. Chem Biol Interact 2003;145:67-75.
- Lebrecht D, Setzer B, Ketelsen UP, Haberstroh J, Walker UA. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation 2003;108:2423-9.
- Lebrecht D, Kokkori A, Ketelsen UP, Setzer B, Walker UA. Tissue-specific mtDNA lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J Pathol 2005;207:436-44.
- Olson RD, Mushlin PS. Doxorubicin cardiotoxicity – analysis of prevailing hypotheses. FASEB J 1990;4:3076-86.
- Quiles JL, Huertas JR, Battino M, Mataix J, Ramirez-Tortosa MC. Antioxidant nutrients and adriamycin toxicity. Toxico-logy 2002;180:79-95.
- Gustafson DL, Swanson JD, Pritsos CA. Modulation of glutathione and glutathione-dependent antioxidant enzymes in mouse heart following doxorubicin therapy. Free Radic Res Commun 1993;19:111-20.
- Johnson WT, Johnson LA, Lukaski HC. Serum superoxide dismutase 3 (extracellular superoxide dismutase) activity is a sensitive indicator of Cu status in rats. J Nutr Biochem 2005;16:682-92.
- Abdel-Wahab MH, El-Mahdy MA, Abd-Ellah MF, Helal GK, Khalifa F, Hamada FMA. Influence of p-coumaric acid on doxorubicin-induced oxidative stress in rat’s heart. Pharmacol Res 2003;48:461-5.