
Natural dietary anti-cancer chemopreventive compounds: redox-mediated differential signaling mechanisms in cytoprotection of normal cells versus cytotoxicity in tumor cells1
Introduction
Many dietary phytochemicals exhibit beneficial effects to health, including the prevention of diseases such as cancer, as well as neurological, cardiovascular, inflammatory, and metabolic diseases. Evolutionarily, herbivorous and omnivorous animals have been ingesting plants. This interaction between “animal–plant” ecosystems has resulted in an elaborate system of detoxification and defense mechanisms evolved by animals including humans. The condition of stress generated by electrophiles or xenobiotics may be referred to as electrophilic stress. Mammalian cells, including human cells, respond to these dietary phytochemicals by a “non-classical receptor sensing” mechanism of electrophilic chemical-stress typified by “thiol-modulated” cellular signaling events primarily leading to the gene expression of pharmacologically beneficial effects, but sometimes unwanted cytotoxicity also. Our laboratory has been studying two groups of dietary phytochemical cancer-chemopreventive compounds (isothiocyanates and polyphenols)[1,2], which are effective in chemical-induced, as well as genetically-induced, animal carcinogenesis models[3,4]. These compounds typically generate “cellular stress” and modulate gene expression of phase II detoxifying/antioxidant enzymes.
Multicellullar organisms rely on highly-organized pathways to orchestrate the many extracellular clues received by the cells and to convert them into specific physiological processes. The classical first step in this cascade of molecular events that are collectively referred to as signal transduction pathways is the specific interaction of an extracellular ligand with its receptor at the cell membrane[5]. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) have been proposed as second messengers in the activation of several signaling pathways leading to mitogenesis or apoptosis[5]. The effective transmission of information requires specificity, and how ROS signaling occurs with specificity and without oxidative damage remains poorly understood[6]. In addition, gene expression responses to oxidative stress are necessary to ensure cell survival and are largely attributed to specific redox-sensitive transcription factors[7]. Redox-sensitive cysteine residues are known to sense and transduce changes in cellular redox status caused by the generation of ROS and the presence of oxidized thiols[8].
Sporn and Liby[9] noted that the principal need in the chemoprevention of cancer remains the discovery of new agents that are effective and safe, and the development of new dose-scheduling paradigms that will allow their beneficial use over relatively long periods of time, but not necessarily constantly, in a manner that is essentially free of undesirable side effects. It has been reported[10] that human cancers overexpressing genes are specific to a variety of normal human tissues, including normal tissues other than those from which the cancer originated, suggesting that this general property of cancer cells plays a major role in determining the behavior of the cancers, including their metastatic potential. Surh[11] observed that the disruption or deregulation of intracellular-signaling cascades often leads to malignant transformation of cells, and it is therefore important to identify the molecules in the signaling network that can be affected by individual chemopreventive phytochemicals to allow for better assessment of their underlying molecular mechanisms. Conney[12] suggested that tailoring the chemopreventive regimen to the individual or to groups of individuals living under different environmental conditions, or with different mechanisms of carcinogenesis, may be an important aspect of cancer chemoprevention in human populations. Given the inability to guarantee the long-term safety of current chemopreventive regimens, Sporn[9] suggested two approaches: the more widespread use of low-dose combinations of chemopreventive agents with the goal of achieving a therapeutic synergy between individual drugs while reducing their individual toxicities, or the use of intermittent, rather than constant, chronic dosing of chemopreventive agents.
In the current review, we will attempt to shed light on redox-mediated signaling translating into gene expression and in vivo pharmacological events. We will explore dietary cancer-chemopreventive phytochemicals, discuss the link between oxidative/electrophilic stress and the redox circuitry, and consider a brief overview of redox-sensitive transcription factors. We will then discuss the kelch-like erythroid Cap’n’Collar (CNC) homologue-associated protein 1 (Keap1)-nuclear factor-E2-related factor 2 (Nrf2) axis in redox signaling, an important target for preventive and possibly therapeutic intervention in many types of cancers. We will also discuss the Nrf2 paradigm in gene expression, transcriptome profiling of disparate gene categories, and the pharmacotoxico-genomic relevance of redox-sensitive Nrf2. Finally, we will discuss the redox regulation of cell death mechanisms.
Natural dietary anti-cancer chemopreventive compounds
Because carcinogenesis comprises three different stages: initiation, promotion, and progression, many potential cancer-protective (chemopreventive) agents can be categorized broadly as blocking agents (which impede the initiation stage) or suppressing agents (which arrest or reverse the promotion and progression of cancer), presumably by affecting or perturbing crucial factors that control cell proliferation, differentiation, senescence, or apoptosis[2,13]. Dietary chemopreventive compounds functioning as detoxifying enzyme inducers primarily include phenolic and sulfur-containing compounds. Phenolic compounds may be classified into polyphenols and flavonoids, whereas sulfur-containing compounds may be classified into isothiocyanates and organosulfur compounds[1]. Representative examples of polyphenols include epigallocatechin-3-gallate (EGCG) from green tea, curcumin from turmeric, and resveratrol from grapes; whereas flavonoids are exemplified by quercetin from citrus fruits and genistein from soy. Isothiocyanates include, amongst others, sulforaphane (SFN) from broccoli, phenethyl isothiocyanate (PEITC) from turnips and watercress, and allyl isothiocyanate from brussels sprout. Organosulfur compounds chiefly include diallyl sulfides from garlic oil. Dietary isothiocyanates are derived in vivo from the hydrolysis of glucosinolates present in cruciferous vegetables.
Our laboratory has worked extensively towards understanding the molecular mechanisms in vitro and determining the chemopreventive efficacy in vivo of polyphenols (eg EGCG and curcumin), and isothiocyanates (eg SFN and PEITC), and combinations of these phytochemicals to elicit maximum efficacy in cancer cells/tumorigenic tissue with minimum toxicity to normal cells. It is indeed quite puzzling as to how these compounds can differentiate between “normal” versus “abnormal tumor” cells in terms of signaling, gene expression, and pharmacological effects. We have shown[14] that EGCG treatment in human colon HT-29 cancer cells causes damage to mitochondria, and that c-Jun N-terminal kinase (JNK) mediates EGCG-induced apoptotic cell death. Similarly, it was shown[15] that PEITC can induce apoptosis in HT-29 cells in a time- and dose-dependent manner via the mitochondrial caspase cascade, and that the “activation” of JNK appears to be critical for the initiation of the apoptotic processes. On the other hand, EGCG, curcumin, SFN, and PEITC appear to “inhibit” (instead of activate) lipopolysaccharide (LPS)-induced NF-κB activation in the same cell type, HT-29 cells, stably transfected with an NF-κB luciferase reporter construct[16]. In addition, the mechanism of action of SFN was recently[17] attributed to the inhibition of p38 mitogen-activated protein kinase (MAPK) isoforms which contributed to the induction of antioxidant response element (ARE)-mediated heme oxygenase-1 (HO-1) gene expression in human hepatoma HepG2 cells. Extracellular signal-regulated protein kinase (ERK) and JNK pathways were activated[18] by PEITC treatment in human prostate cancer PC-3 cells. Interestingly, a combination of PEITC and curcumin was found to have an additive effect[19] on the induction of apoptosis in PC-3 cells stably transfected with an NF-κB luciferase reporter construct and involved an inhibition of epidermal growth factor receptor (EGFR), its downstream signaling including PI3K and Akt. In vivo, PEITC and curcumin alone or in combination exhibited significant cancer-preventive activities in NCr immunodeficient (nu/nu) mice bearing subcutaneous xenografts of PC-3 cells[20]. It is tempting to speculate that “abnormal tumor” cells such as PC-3 and HT-29 cells require the overexpressed or hyper-activated NF-κB and/or EGFR for cell survival/proliferation, whereas “normal” cells do not require these signaling molecules to do so. The blockade of NF-κB and/or EGFR signaling by these compounds would sensitize tumor cells to die, but not the normal cells (but, instead, in normal cells these compounds will redox-dependently affect the Nrf2-mediated cellular detoxifying/antioxidant defense enzymes, which will be discussed in greater detail later on), and hence the potential specificities between “abnormal tumor” versus “normal” cells. Recently, we[3] and others[21] showed that SFN inhibited adenoma formation in the gastrointestinal tract of genetically mutant ApcMin/+ mice, and that the concentrations of SFN and its metabolite SFN– glutathione (GSH) were found to be between 3 and 30 nmol/g (~3–30 µmol/L), which resembled that of the in vitro cell culture systems[22]. Interestingly, under such conditions, using immunohistochemical staining of the adenomas indicated that SFN significantly suppressed the expression of phosphorylated-JNK, phosphorylated-ERK and phosphorylated-Akt, which were found to be highly expressed in the adenomas of ApcMin/+ mice versus normal mucosa[3]. When the acute effect of SFN on the gene expression profiles was investigated in the small intestine polyps of the SFN-treated Apc+/Min mice by using Affymetrix microarray platforms, the results showed that genes involved in apoptosis, cell growth, and maintenance, rather than phase II detoxifying genes, were modulated in the polyps of the SFN-treated Apc/Min+ mice. Some of the pro-apoptotic genes such as MBD4 and serine/threonine kinase 17b, TNF receptor superfamily member 7 and TNF (ligand) superfamily member 11 were upregulated, while some pro-survival genes such as cyclin-D2, integrin β1, and Wnt 9A were significantly downregulated in adenomas treated with SFN. Importantly, two important genes involved in colorectal carcinogenesis, arachidonate 15-lipoxygenase (15-LOX) and COX-2 were found to be increased and decreased respectively by SFN[3]. Most recently, we found that in the colon of the Nrf2 –/– mice challenged with the classical inflammatory agent dextran sodium sulfate, the inflammatory proteins such as COX-2 and iNOS were overexpressed with a concomitant decreased expression of cellular antioxidant enzymes such as HO-1 and NQO1[23]. These results imply that the induction of antioxidant enzymes by dietary cancer chemopreventive compounds can potentially counteract the oxidative/inflammatory signaling pathways as observed by Dinkova-Kostova et al[24].
Redox-mediated signaling
Oxidative stress and redox circuitry Although oxidants are constantly generated for essential biologic functions, excess generation or an imbalance between oxidants and antioxidants can produce a common pathophysiological condition known as oxidative stress[25]. Mediators of oxidative stress, that is, ROS, also function as second messengers in signal transduction. In the light of this knowledge, oxidative stress has been remarkably defined as perturbations in redox circuitry[6]. Indeed, proteins present on cell surfaces and located in extracellular fluids undergo oxidation in diverse pathophysiological conditions, and a growing body of evidence suggests that the steady-state oxidation is responsive to diet[25].
Interestingly, redox compartmentation functions as a mechanism for specificity in redox signaling and oxidative stress, with the relative redox states[6] from most reducing to most oxidizing being mitochondria>nuclei>cytoplasm>endoplasmic reticulum>extracellular space. Hansen et al[6] noted that a circuitry model for redox signaling and control has been developed based on the observations that three major thiol/disulfide couples, namely glutathione (GSH)/glutathione disulfide (GSSG), reduced thioredoxin [Trx-(SH)2]/oxidized thioredoxin (Trx-SS), and cysteine (Cys)/cystine (CySS) are not in redox equilibrium and therefore can function as control nodes for many different redox-sensitive processes. In this model, redox switches and pathways exist in parallel circuits, with electron flow from NADPH as a central electron donor to ROS and O2 as electron acceptors.
Given the involvement of ROS and RNS in a multiplicity of physiological responses through the modulation of signaling pathways, studies on reactive oxygen and nitrogen species (RONS) signaling have received considerable attention in recent times. Forman[5] noted primarily that: (i) antioxidant enzymes are essential “turn-off” components in signaling; (ii) spatial relationships are probably more important in RONS signaling than the overall “redox state” of the cell; and (iii) deprotonation of the cysteine to form the thiolate, which can react with RONS, occurs in specific protein sites providing specificity in signaling. The bacterial transcription factor OxyR[8] mediates the cellular response to both ROS and RNS by controlling the expression of the OxyR regulon, which encodes proteins involved in the H2O2 metabolism and the cytoplasmic thiol redox response. In budding yeast, the transcription factor Yap1 (yeast AP1), which is a basic leucine zipper (bZip) transcription factor, confers the cellular response to redox stress by controlling the expression of the regulon that encodes most yeast antioxidant proteins[8]. Interestingly, Georgiou[26] noted that in both bacteria and yeast, the redox control networks exemplified by OxyR and Yap1, respectively, exhibit conserved dynamic features, namely autoregulation (which in yeast is accomplished via the reduction of Yap1 by thioredoxin) and hysteresis. Indeed, the question of whether these features are intrinsic properties of the regulatory architecture required for proper adaptation to redox stress remains to be resolved. Jacob[27] observed that disulfide-S-oxides are formed from glutathione under oxidizing conditions and specifically modulate the redox status of thiols, indicating the existence of specialized cellular oxidative pathways. This supports the paradigm of oxidative signal transduction and provides a general pathway whereby ROS can convert thiols into disulfides.
Redox-sensitive transcription factors Many transcription factors are redox-sensitive, including activator protein-1 (AP-1), NF-κB, Nrf2, p53, and the glucocorticoid receptor[6]. Such sensitivity involves at least 2 redox-sensitive steps, one in the activation of the signaling cascade and another in DNA binding, and possibly additional redox-sensitive nuclear processes such as nuclear import and export[6]. Nuclear and cytoplasmic redox couples perform distinct functions during redox-sensitive transcription factor regulation.
AP-1 is responsive to low levels of oxidants resulting in AP-1/DNA binding and an increase in gene expression. AP-1 activation is due to the induction of JNK activity by oxidants resulting in the phosphorylation of serine 63 and serine 73 in the c-Jun transactivation domain[7,28,29]. With high concentrations of oxidants, AP-1 is inhibited and gene expression is impeded. The inhibition of AP-1/DNA interactions is attributed to the oxidation of specific cysteine residues in c-Jun’s DNA binding region, namely cysteine 252[7,30]. Xanthoudakis and Curran[31] reported that a DNA repair enzyme apurinic/apyrimidinic endonuclease (APE), also termed redox factor-1 (Ref-1), possessed oxidoreductase activity and was responsible for the redox regulation of AP-1. Oxidized AP-1 could be effectively reduced by Ref-1, restoring DNA binding activity. Trx1 was shown to be a critical player in AP-1 regulation by virtue of its ability to reduce oxidized Ref-1[32]. Thus, for AP-1, a nuclear pathway to reduce the Cys of the DNA-binding domain is distinct[6] from the upstream redox events that activate the signaling kinase pathway.
Similarly, NF-κB contains a redox-sensitive critical cysteine residue (cysteine 62) in the p50 subunit that is involved in DNA binding[7,33]. NF-κB is normally sequestered in the cytoplasm by IκB, but under oxidative conditions, IκB is phosphorylated by IκB kinase (IKK), ubiquitinated, and subsequently degraded. ROS production appears to be necessary to initiate the events leading to the dissociation of the NF-κB/IκB complex[6], but excessive ROS production (oxidative stress) results in the oxidation of cysteine 62 which does not affect its translocation to the nucleus, but rather, interferes with DNA binding and decreases gene expression[7,34]. Overexpression of Trx1 inhibited NF-κB activity, but overexpression of nuclear-targeted Trx1 enhanced NF-κB activity[35]. These findings suggested[6] that Trx1 plays distinct roles within the cytoplasm (regulation of activation and dissociation of IκB), and within the nucleus (regulation of DNA binding).
Pivotal to the antioxidant response[36–39] typical in mammalian homeostasis and oxidative stress is the important transcription factor Nrf2 that has been extensively studied by many research groups[1,36–43]. Nrf2 is indispensable to cellular defense against many chemical insults of endogenous and exogenous origin, which play major roles in the etiopathogenesis of many cancers and inflammation-related diseases such as inflammatory bowel disease[23] and Parkinson’s disease[44]. The role of Nrf2 as a critical redox-sensitive transcription factor will be discussed in greater detail in the following section devoted to redox signaling with specific emphasis on the Keap1–Nrf2 axis.
Keap1–Nrf2 axis in redox signaling Under homeostatic conditions, Nrf2 is mainly sequestered in the cytoplasm by a cytoskeleton-binding protein called Keap1[40,45,46]. Zhang and Hannink[47] have identified 2 critical cysteine residues in Keap1, namely C273 and C288, that are required for Keap1-dependent ubiquitination of Nrf2 as shown in Figure 1. They have also identified[47] a third cysteine residue in Keap1, namely C151, that is uniquely required for the inhibition of Keap1-dependent degradation of Nrf2 (Figure 1) by sulfo-raphane and oxidative stress. It has also been shown[48] that Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex that controls steady-state levels of Nrf2 in response to cancer-preventive compounds and oxidative stress. Moreover, it has been report-ed[49] that Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via an active Crm1/exportin-dependent nuclear export mechanism. When challenged with oxidative stress, Nrf2 is quickly released from Keap1 retention and translocates to the nucleus[40,50]. We have recently identified[40] a canonical redox-insensitive nuclear export signal (NES) (537LKKQLSTLYL546) located in the leucine zipper (ZIP) domain of the Nrf2 protein, as well as a redox-sensitive NES (173LLSI-PELQCLNI186) in the transactivation (TA) domain of Nrf2[51]. Once in the nucleus, Nrf2 not only binds to the specific consensus cis-element called ARE present in the promoter region of many cytoprotective genes[43,46], but also to other trans-acting factors such as small Maf (MafG and MafK)[52] that can coordinately regulate gene transcription with Nrf2. We have also reported[41] that different segments of the Nrf2 transactivation domain have different transactivation potential and that different MAPK have differential effects on Nrf2 transcriptional activity with ERK and JNK pathways playing an unequivocal role in the positive regulation of Nrf2 transactivation domain activity[41]. To better understand the biological basis of signaling through Nrf2, it has also become imperative to identify possible interacting partners of Nrf2 such as co-activators or co-repressors apart from trans-acting factors such as small Maf[52,53].
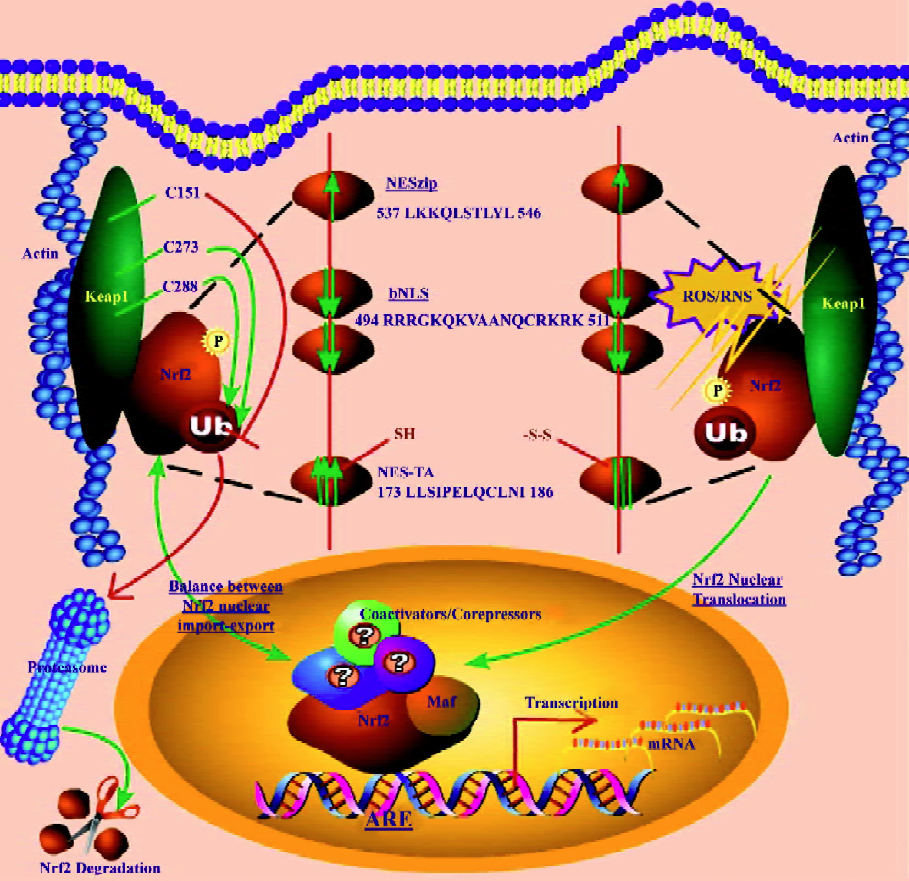
A salient feature of the canonical redox-insensitive Nrf2–NES (NESzip, 537LKKQLSTLYL546) alluded to earlier, is its overlap with the ZIP domain. This overlap implies that when Nrf2 forms heterodimers via its ZIP with other bZIP proteins, such as its obligatory binding partner small MafG/K proteins, the NES motif may be simultaneously masked[40]. Further, we observed[40] that Nrf2 heterodimerization via ZIP with MafG/K may not only enhance the DNA binding specificity of Nrf2, but may also effectively recruit Nrf2 into the nucleus by simultaneously masking the NES activity. Accumulating evidence shows that after fulfilling its transactivation function, Nrf2 is destined for proteasomal degradation in the cytoplasm, although some weak degradation activity may also exist within the nucleus. Hence, Nrf2 signaling may be turned on and off rapidly to match the rapid changes of the cellular redox status[40]. Interestingly, a bipartite nuclear localization signal (bNLS, 494RRRGKQKVAANQCRKRK511) as well as an NES (545LKRRLSTLYL554) have been identified[54] in the C terminus of Nrf2, further underscoring the importance of nuclear import and export in controlling the subcellular localization of Nrf2.
Unlike the NESzip, the new functional redox-sensitive NES located in the transactivation (TA) domain of Nrf2 (NESTA, 173LLSIPELQCLNI186) possesses a reactive cysteine residue (C183)[51] and exhibits a dose-dependent nuclear translocation when treated with sulforaphane[51]. The NESTA motif functions as a redox-sensitive switch that can be turned on/off by oxidative signals and determines the subcellular localization of Nrf2[51]. These discoveries suggest that Nrf2 may be able to transduce redox signals in a Keap-1-independent manner; however, Keap1 may provide additional regulation of Nrf2 both in basal and inducible conditions.
Li et al[51] have recently proposed a new model for Nrf2 redox signaling. As depicted in Figure 1, the Nrf2 molecule possesses multivalent NES/NLS motifs, and their relative driving forces are represented by the size and direction of the arrows. Under unstimulated conditions (to the left of Figure 1), the combined nuclear-exporting forces of NESTA[51] and NESzip[51] may counteract the nuclear importing force of the bNLS[54]. As a result, Nrf2 exhibits a predominantly whole cell distribution. While the majority of Nrf2 molecules remain in the cytoplasm, the residual nuclear Nrf2 may account for the basal or constitutive Nrf2 activities. The observation of a small percentage of cells exhibiting nuclear and cytosolic distribution of Nrf2 may reflect the hyper- and hypo-oxidative condition of individual cells, respectively. When challenged with oxidative stress (to the right of Figure 1), the redox-sensitive NESTA is disabled, but the redox-insensitive NESzip remains functional[40,51], and the bNLS motif may remain functionally uninterrupted[51,55]. Since the driving force of the NESzip is weaker than that of the bipartite bNLS, the nuclear importing force mediated by the bNLS prevails and triggers Nrf2 nuclear translocation[51] as shown in Figure 1.
Gene expression and in vivo pharmacological effects
Nrf2 paradigm in gene expression Nrf2 knockout mice are greatly predisposed to chemical-induced DNA damage and exhibit higher susceptibility towards cancer development in several models of chemical carcinogenesis[43]. Observations that Nrf2-deficient mice are refractory to the protective actions of some chemopreventive agents[43] highlight the importance of the Keap1–Nrf2–ARE signaling pathway as a molecular target for prevention. Hence, several studies have focused on using these Nrf2-deficient mice for pharma-cogenomic or toxicogenomic profiling of the transcrip-tome in response to dietary chemopreventive agents[53,56–59] and toxicants[60].
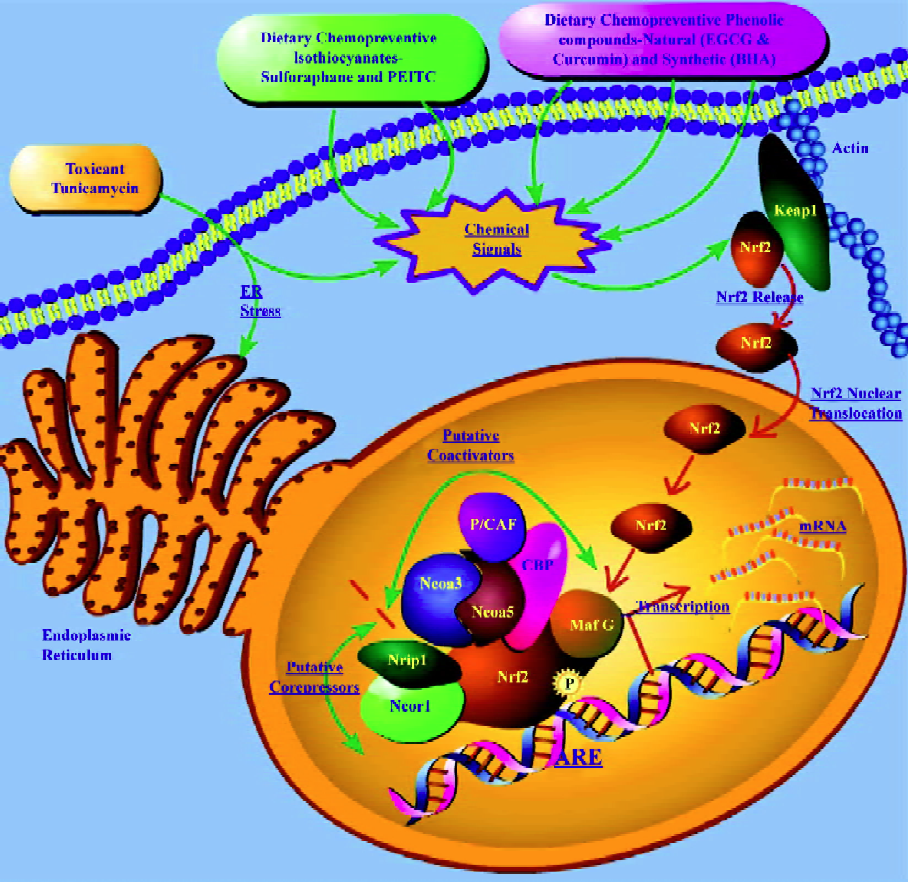
Transcriptome profiling of putative Nrf2 co-activators and co-repressors In recent times, there has been renewed interest in dissecting the interacting partners of Nrf2, such as co-activators and co-repressors, which are co-regulated with Nrf2 to better understand the biochemistry of Nrf2. In a recent microarray study[56] with Nrf2 knockout mice, it was reported that the CREB-binding protein (CBP) was upregul-ated in mice liver on treatment with EGCG in an Nrf2-dependent manner. Katoh et al[61] showed that 2 domains of Nrf2 (Neh4 and Neh5) cooperatively bind CBP and synergistically activate transcription. It was also demonstrated[41] previously, using a Gal4-Luc reporter co-transfection assay system in HepG2 cells, that the nuclear transcriptional co-activator CBP, which can bind to the Nrf2 transactivation domain and can be activated by the ERK cascade, showed synergistic stimulation with Raf on the transactivation activities of both the chimera Gal4-Nrf2 (1-370) and the full-length Nrf2. In a recent microarray study with butylated hydroxyanisole (BHA) treatment in Nrf2 knockout mice[53], we observed the upregulation of the trans-acting factor v-maf musculoaponeurotic fibrosarcoma oncogene family protein G avian (MafG), nuclear receptor co-repressor 1 (Ncor1) and nuclear receptor co-repressor interacting protein (Nrip1) as well as the downregulation of the nuclear receptor co-activator 3 (Ncoa3) in an Nrf2-dependent manner. Similarly, in another transcriptome profiling study in Nrf2 knockout mice with the endoplasmic reticulum (ER) stress inducer tunicamycin[60], that modulates the unfolded protein response (UPR), we observed the upregulation of the P300/CBP-associated factor (P/CAF), trans-acting factor v-maf musculoaponeurotic fibrosarcoma oncogene family, protein F (MafF), nuclear receptor co-activator 5 (Ncoa5), Smad nuclear interacting protein (Snip1) and Nrip1 as well as downregulation of the src family associated phosphoprotein 2 (Scap2) in an Nrf2-dependent manner. Although microarray expression profiling cannot provide evidence of binding between partners, this could potentially suggest[53] that co-repressors Ncor1 and Nrip1 and co-activator Ncoa3, such as CBP in the previous studies, may serve as putative BHA-regulated nuclear interacting partners of Nrf2 in eliciting the cancer chemopreventive effects of BHA, and that co-repressor Nrip1 and co-activators P/CAF and Ncoa5 may serve as putative tunicamycin-regulated nuclear interacting partners of Nrf2 in eliciting the UPR-responsive events[60] in vivo. Furthermore, the induction of Nrip1 was observed in both the small intestine and liver with BHA suggesting that the Nrf2/ARE pathway may play an important role in BHA-elicited regulation of this gene. It has also been shown recently[62] that the co-activator P/CAF could transcriptionally activate a chimeric Gal4-Nrf2-luciferase system containing the Nrf2 transactivation domain in HepG2 cells. In addition, P/CAF, which is known[63] to be a histoneacetyl transferase protein, has recently been shown[64] to mediate DNA damage-dependent acetylation on most promoters of genes involved in the DNA-damage and ER-stress response, which validates the observation[60] of P/CAF induction via Nrf2 in response to tunicamycin-induced ER stress.
Taken together, it is tempting to speculate that dietary chemopreventive agents such as BHA and EGCG, and toxicants such as tunicamycin, may elicit their chemopreventive and pharmacological/toxicological events through the ARE by means of a multi-molecular complex[53,60] of co-activators and co-repressors that function in concert with the redox-sensitive transcription factor Nrf2 as depicted in Figure 2. The putative multi-molecular complex may involve Nrf2 along with the transcriptional co-repressors Ncor1 and Nrip1, and the transcriptional co-activators Ncoa3, Ncoa5, P/CAF, and CBP, in addition to the currently known trans-acting factors such as MafG[52], with multiple interactions between the members of the putative complex as has been shown recently with the p160 family of proteins[62]. As shown in Figure 2, chemical signals generated by dietary chemopreventive agents or toxicants may cause Nrf2 nuclear translocation that sets in motion a dynamic machinery of co-activators and co-repressors that may form a multi-molecular complex with Nrf2 to modulate transcriptional response through the ARE. Further studies of a biochemical nature would be needed to substantiate this hypothesis and extend our current understanding of Nrf2 regulation in chemoprevention with BHA or EGCG and in tunicamycin-mediated ER stress.
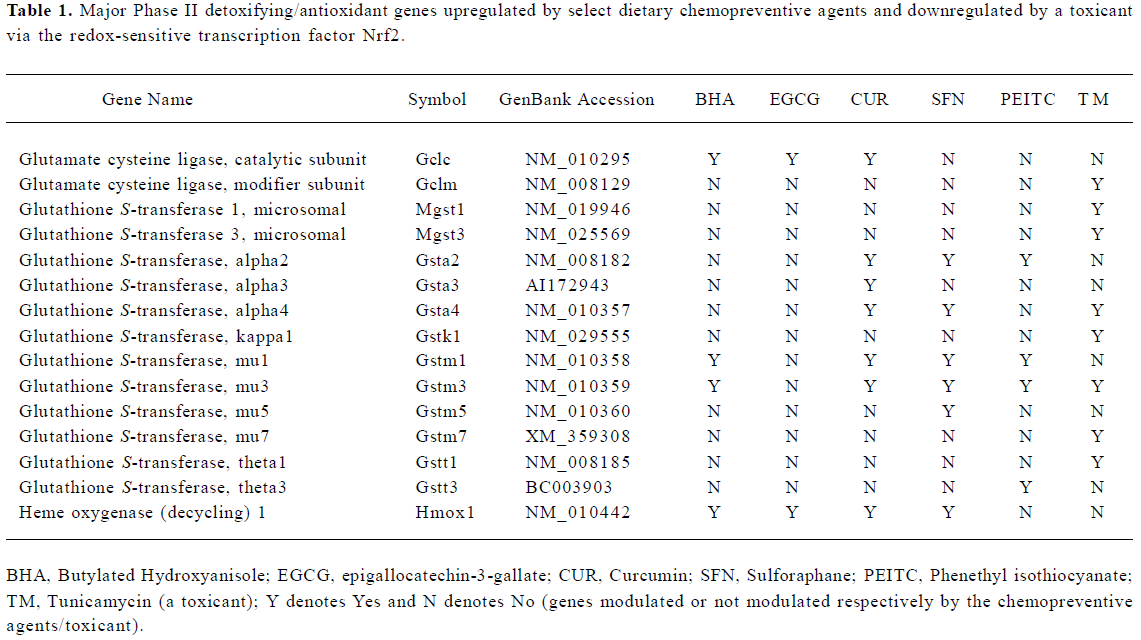
Coordinated regulation of phase I, II, and III drug metabolizing enzyme/transporter genes via Nrf2 Dietary chemopreventives such as BHA, EGCG, and curcumin could coordinately regulate the phase I, II, and III xenobiotic metabolizing enzyme genes as well as antioxidative stress genes through Nrf2-dependent pathways in vivo[53,56,57]. Interes-tingly, a coordinated response involving phase I, II, and III genes was also observed in vivo on ER stress induction with the toxicant tunicamycin in an Nrf2-dependent manner[60]. The array of genes coordinately[53] modulated included phase I drug metabolizing enzymes, phase II detoxification and antioxidant genes, and phase III transporter genes. The regulation of these genes could have significant effects on the prevention of tumor initiation by enhancing the cellular defense system, preventing the activation of procarcinogens/reactive intermediates, and increasing the excretion/efflux of reactive carcinogens or metabolites[53,57]. Indeed, the induction of phase II enzymes itself achieves protection against the toxic and neoplastic effects of many carcinogens. In addition to enzymes (that conjugate to functionalized xenobiotics) such as glutathione S-transferases and UDP-glucuronosyltransferases, a provisional and partial list of phase II genes might include[65] NAD(P)H:quinone reductase, epoxide hydrolase, dihydrodiol dehydrogenase, gamma-glutamylcysteine synthetase, heme oxygenase-1, leukotriene B4 dehydrogenase, aflatoxin B1 dehydrogenase, and ferritin. The major phase II genes modulated via Nrf2 by BHA, EGCG, curcumin, sulforaphane, PEITC, and tunicamycin are summarized in Table 1.
Full table
Spatial and temporal control of Nrf2-mediated gene expression It has been observed that the regulation of Nrf2-mediated gene expression takes place at both spatial and temporal levels in in vivo whole animals. For example, the dietary chemopreventive BHA did not modulate the transcription of NAD(P)H:quinine oxidoreductase (NQO1) at 3 h; however, NQO1 was induced by BHA at 12 h[53]. The relatively delayed induction of the NQO1 gene compared with the other phase II genes in response to BHA points to the possibility of differential kinetics of BHA-regulated phase II gene response with temporal or time-dependent specificity. However, many other genes were modulated differentially at 3 h in response to BHA between the small intestine and liver[53] indicating certain spatial or tissue/compartment-dependent control of gene expression. Similar phenomena were also observed in microarray studies with EGCG[56], curcumin[57], SFN[58], and PEITC[59]. This could be attributed to a complex of physiological factors including partitioning across the gastrointestinal tract, intestinal transit time, uptake into the hepatobiliary circulation, exposure parameters such as Cmax, Tmax and, AUC, and the pharmacokinetics of disposition after oral administration[53]. Other potential complicating factors could be the possibility of the differential tissue/organ-dependent expression of endogenous Keap1 and Nrf2 in conjunction with other tissue-specific/general nuclear co-regulators and co-repressors[41,62,66] which could dynamically shift the equilibrium locally in favor of or against Nrf2-mediated transcription of different ARE-driven genes.
Pharmacotoxicogenomic relevance of redox-sensitive Nrf2 Recently, it was reported[67] that Nrf1, another member of the CNC family of basic ZIP proteins that is structurally similar to Nrf2, is normally targeted to the ER membrane, and that ER stress induced by tunicamycin in vitro may play a role in modulating Nrf1 function as a transcriptional activator. As a protein-folding compartment, the ER is exquisitely sensitive to alterations in homeostasis, and provides stringent quality control systems to ensure that only correctly folded proteins transit to the Golgi and unfolded or misfolded proteins are retained and ultimately degraded. A number of biochemical and physiological stimuli, including perturbation in redox status, can disrupt ER homeostasis and impose stress to the ER, subsequently leading to accumulation of unfolded or misfolded proteins in the ER lumen[68]. The ER has evolved highly specific signaling pathways called the UPR to cope with the accumulation of unfolded or misfolded proteins[68,69]. ER stress stimulus by thapsigargin has also been shown[70] to activate the c-JNK or stress-activated protein kinase that is a member of the MAPK cascade[71]. More-over, it has been reported that the coupling of ER stress to JNK activation involves the transmembrane protein kinase IRE1 by binding to an adaptor protein TRAF2, and that IRE1α–/– fibroblasts were impaired in JNK activation by ER stress[72]. It has been previously reported that PEITC, contained in large amounts in cruciferous vegetables such as watercress, activates JNK1[73], and that the activation of the ARE by PEITC involves both Nrf2 and JNK1[42] in HeLa cells. It has also been demonstrated that ERK and JNK pathways play an unequivocal role in the positive regulation of Nrf2 transactivation domain activity in vitro in HepG2 cells. Although there is growing interest amongst researchers in targeting the UPR against cancerous tumor growth[74], there is currently little understanding of the role of Nrf2 in modulating the UPR in vivo. Interestingly, in a Nrf2-deficient murine model[60], glutathione peroxidase 3 (Gpx3) is upregulated and superoxide dismutase 1 (Sod1) is downregulated by tunica-mycin in an Nrf2-deficient manner, which can have important implications in oxidative stress-mediated[22] pathophy-siology or ER stress caused by perturbations in redox circuitry[22,68,75]. The identification of novel molecular targets that are regulated by the toxicant tunicamycin via Nrf2 in vivo raises possibilities for targeting the UPR proteins in future to augment or suppress the ER stress response and modulate disease progression. Future toxicogenomic/toxicokinetic studies may provide new biological insights into the diverse cellular and physiological processes that may be regulated by the UPR signaling pathways in cancer pharmacology and toxicology and the role (s) of Nrf2 in these processes.
Redox regulation of cellular signaling molecules leading to cell death mechanisms It has been demonstrated[76] that the redox regulation of platelet-derived growth factor receptor (PDGFr) tyrosine autophosphorylation and its signaling are related to NADPH oxidase activity through protein kinase C (PKC) and phosphoinositide-3-kinase (PI3K) activation and H2O2 production. Cheng and Liu[77] reported that lead (Pb) increased LPS-induced liver damage in rats by modulation of TNF-alpha and oxidative stress through PKC and p42/44 MAPK. Recently, it was shown[78] that Rac upregulated the tissue inhibitor of metalloproteinase-1 expression by redox-dependent activation of ERK signaling. It has been reported[79] that cancer chemoprevention by the nitroxide antioxidant tempol acts partially via the p53 tumor suppressor.
Trachootham et al[80] have recently demonstrated a selective killing of oncogenically transformed cells by PEITC via a ROS-mediated mechanism. Oncogenic transformation of ovarian epithelial cells with H-Ras(V12) or expression of Bcr-Abl in hematopoietic cells caused elevated ROS generation and rendered the malignant cells highly sensitive to PEITC, which effectively disabled the glutathione antioxidant system and caused severe ROS accumulation preferentially in the transformed cells due to their active ROS output resulting in oxidative mitochondrial damage, inactivation of redox-sensitive molecules, and massive cell death[80]. Xu et al[81] demonstrated an inhibition by SFN and PEITC on NF-κB transcriptional activation as well as downregulation of NF-κB-regulated VEGF, cyclin D1, and Bcl-X(L) gene expression primarily through the inhibition of IKK-beta phos-phorylation, and the inhibition of IκB-alpha phosphorylation and degradation, and decrease of nuclear translocation of p65 in human prostate cancer PC-3 cells. Furthermore, in the same PC-3 cells, PEITC and curcumin substantially inhibited EGFR, PI3K as well as Akt activation/phosphoryla-tion, and the combination of PEITC and curcumin was more effective in the inhibition[19]. These inhibitions can also be recapitulated in vivo in PC-3 transplanted athymic nude mice[20]. Shen and Pervaiz[82] have noted a role for redox-dependent execution in TNF receptor superfamily-induced cell death. Moreover, oxidation of the intracellular environment of hepatocytes by ROS or redox-modulating agents has been recently shown[83] to sensitize hepatocytes to TNF-induced apoptosis that seems to occur only in a certain redox range (in which redox changes can inhibit NF-κB activity, but not completely inhibit caspase activity) by interfering with NF-κB signaling pathways. Nitric oxide has been reported[84] to induce thioredoxin-1 nuclear translocation that may be associated with the p21Ras survival path-way. Also, the p53-independent G1 cell cycle arrest of HT-29 human colon carcinoma cells by SFN was shown[85] to be associated with the induction of p21CIP1 and the inhibition of expression of cyclin D1. A small dose (10 µmol/L) of hydrogen peroxide has been shown[86] to enhance TNF-alpha-induced cell apoptosis, upregulate Bax, and downregulate Bcl-2 expression in the human vascular endothelial cell line ECV304.
Recently, a new functional class of redox-reactive thalidomide analogs with distinct and selective antileukemic activity has been identified[87]. These agents activate the NF of activated T-cells transcriptional pathways while simultaneously repressing NF-κB via a rapid intracellular amplification of ROS that is associated with caspase-independent cell death[87]. This cytotoxicity is highly selective for transformed lymphoid cells and preferentially targets cells in the S-phase of the cell cycle[87]. Capsaicin has been shown[88] to selectively induce apoptosis in mitogen-activated or transformed T cells with rapid increase of ROS; however, neither production of ROS nor apoptosis is detected in capsaicin-treated resting T cells suggesting differential signaling between activated/transformed T cells versus resting T cells. As noted elsewhere, a combination of PEITC and curcumin has been reported[19] to suppress EGFR phosphorylation as well as the phosphorylation of IκB-alpha and Akt. Furthermore, Kim et al[22] reported that SFN at low concentrations showed strong induction of phase II genes that could potentially protect cells from cytotoxic effects; however, at high concentrations (above 50 µmol/L), SFN induced cell death with activation of caspase 3 and JNK1/2, which could be blocked by exogenous glutathione. The extent of SFN-induced cellular stress may thus decide the direction of signaling between cell survival and cell death[22]. The formation of mixed disulfides with GSH, which is known as S-glutathionylation, is a post-translational modification that is emerging as an important mode of redox signaling[89]. Indeed, redox-reactive compounds provide a new tool through which selective cellular properties of redox status and intracellular bioactivation of potential redox-sensitive molecular targets can be leveraged by rational combinatorial therapeutic strategies and appropriate drug design to exploit cell-specific vulnerabilities for maximum drug efficacy[87].
Modulation of apoptosis and cell-cycle control genes via Nrf2 Several genes related to apoptosis and cell-cycle control that are critical in the etiopathogenesis of many cancers have been shown[53,56–60] to be regulated through Nrf2 in response to several dietary chemopreventive agents and toxicants, far too many to be discussed here, at least in normal murine tissues. The reader is referred to references[53,56–60] for a comprehensive list of these genes. Future studies would ascertain whether Nrf2 would be required for the regulation of these apoptosis and cell-cycle control genes in tumor tissues/cells.
Conclusions
In summary, electrophilic stress and oxidative stress, or perturbations in redox circuitry can have a profound influence on the direction of signaling between cell survival and cell death. The potential involvement of cellular redox-sensitive transcription factors can modulate gene expression events and pharmacotoxicologic responses in diseases like cancer, thus providing new molecular targets for preventive or therapeutic intervention. Controlled nutritional studies in future may specifically utilize extracellular redox measurements to explore mechanistic links between diet, health status, and diseases[25]. Taken together, redox-mediated signaling is an important mechanism for the cancer chemopreventive effects elicited by dietary anticancer chemopreventive compounds. Future studies will likely provide additional insights into the intricacies of the redox paradigm in signal transduction pathways with respect to the etiopathogenesis of cancer and its potential for chemopreventive interven-tion.
Acknowledgments
The authors are grateful to all members of the Kong Laboratory as well as researchers in the scientific community whose works have been cited here. The authors also regret the inability to include many other excellent relevant citations in the interests of comprehensive abridgement.
References
- Chen C, Kong AN. Dietary chemopreventive compounds and ARE/EpRE signaling. Free Radic Biol Med 2004;36:1505-16.
- Chen C, Kong AN. Dietary cancer-chemopreventive compounds: from signaling and gene expression to pharmacological effects. Trends Pharmacol Sci 2005;26:318-26.
- Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C, et al. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis 2006;27:2038-46.
- Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res 2006;66:8293-6.
- Forman HJ, Torres M, Fukuto J. Redox signaling. Mol Cell Biochem 2002;234–5:49-62.
- Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol 2006;46:215-34.
- Hansen JM, Watson WH, Jones DP. Compartmentation of Nrf-2 redox control: regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol Sci 2004;82:308-17.
- Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Curr Opin Struct Biol 2004;14:679-86.
- Sporn MB, Liby KT. Cancer chemoprevention: scientific promise, clinical uncertainty. Nat Clin Pract Oncol 2005;2:518-25.
- Lotem J, Netanely D, Domany E, Sachs L. Human cancers overexpress genes that are specific to a variety of normal human tissues. Proc Natl Acad Sci USA 2005; 102: 18 556–61.
- Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer 2003;3:768-80.
- Conney AH. Tailoring cancer chemoprevention regimens to the individual. J Cell Biochem 2004;91:277-86.
- Chemoprevention Working Group. Prevention of cancer in the next millennium: Report of the Chemoprevention Working Group to the American Association for Cancer Research. Cancer Res 1999;59:4743-58.
- Chen C, Shen G, Hebbar V, Hu R, Owuor ED, Kong AN. Epigal-locatechin-3-gallate-induced stress signals in HT-29 human colon adenocarcinoma cells. Carcinogenesis 2003;24:1369-78.
- Hu R, Kim BR, Chen C, Hebbar V, Kong AN. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis 2003;24:1361-7.
- Jeong WS, Kim IW, Hu R, Kong AN. Modulatory properties of various natural chemopreventive agents on the activation of NF-kappaB signaling pathway. Pharm Res 2004;21:661-70.
- Keum YS, Yu S, Chang PP, Yuan X, Kim JH, Xu C, et al. Mechanism of action of sulforaphane: inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res 2006;66:8804-13.
- Xu C, Yuan X, Pan Z, Shen G, Kim JH, Yu S, et al. Mechanism of action of isothiocyanates: the induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Mol Cancer Ther 2006;5:1918-26.
- Kim JH, Xu C, Keum YS, Reddy B, Conney A, Kong AN. Inhibition of EGFR signaling in human prostate cancer PC-3 cells by combination treatment with beta-phenylethyl isothiocyanate and curcumin. Carcinogenesis 2006;27:475-82.
- Khor TO, Keum YS, Lin W, Kim JH, Hu R, Shen G, et al. Combined inhibitory effects of curcumin and phenethyl isothiocyanate on the growth of human PC-3 prostate xenografts in immunodeficient mice. Cancer Res 2006;66:613-21.
- Myzak MC, Dashwood WM, Orner GA, Ho E, Dashwood RH. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. Faseb J 2006;20:506-8.
- Kim BR, Hu R, Keum YS, Hebbar V, Shen G, Nair SS, et al. Effects of glutathione on antioxidant response element-mediated gene expression and apoptosis elicited by sulforaphane. Cancer Res 2003;63:7520-5.
- Khor TO, Huang MT, Kwon KH, Reddy BS, Kong AN. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res 2006;66:11580-4.
- Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, Suh N, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci USA 2005;102:4584-9.
- Moriarty-Craige SE, Jones DP. Extracellular thiols and thiol/disulfide redox in metabolism. Annu Rev Nutr 2004;24:481-509.
- Georgiou G. How to flip the (redox) switch. Cell 2002;111:607-10.
- Jacob C, Lancaster JR, Giles GI. Reactive sulphur species in oxidative signal transduction. Biochem Soc Trans 2004;32:1015-7.
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem 1995; 270: 16 483–6.
- Smeal T, Binetruy B, Mercola D, Grover-Bardwick A, Heidecker G, Rapp UR, et al. Oncoprotein-mediated signalling cascade stimulates c-Jun activity by phosphorylation of serines 63 and 73. Mol Cell Biol 1992;12:3507-13.
- Abate C, Patel L, Rauscher FJ III, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science 1990;249:1157-61.
- Xanthoudakis S, Curran T. Redox regulation of AP-1: a link between transcription factor signaling and DNA repair. Adv Exp Med Biol 1996;387:69-75.
- Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci USA 1997;94:3633-8.
- Matthews JR, Kaszubska W, Turcatti G, Wells TN, Hay RT. Role of cysteine62 in DNA recognition by the P50 subunit of NF-kappa B. Nucleic Acids Res 1993;21:1727-34.
- Galter D, Mihm S, Droge W. Distinct effects of glutathione disulphide on the nuclear transcription factor kappa B and the activator protein-1. Eur J Biochem 1994;221:639-48.
- Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, . Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J Biol Chem 1999; 274: 27 891–7.
- Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 1999; 274: 26 071–8.
- McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, et al. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res 2001;61:3299-307.
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 2002;62:5196-203.
- Prochaska HJ, De Long MJ, Talalay P. On the mechanisms of induction of cancer-protective enzymes: a unifying proposal. Proc Natl Acad Sci USA 1985;82:8232-6.
- Li W, Jain MR, Chen C, Yue X, Hebbar V, Zhou R, . Nrf2 possesses a redox-insensitive nuclear export signal overlapping with the leucine zipper motif. J Biol Chem 2005; 280: 28 430–8.
- Shen G, Hebbar V, Nair S, Xu C, Li W, Lin W, . Regulation of Nrf2 transactivation domain activity. The differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. J Biol Chem 2004; 279: 23 052–60.
- Keum YS, Owuor ED, Kim BR, Hu R, Kong AN. Involvement of Nrf2 and JNK1 in the activation of antioxidant responsive element (ARE) by chemopreventive agent phenethyl isothiocyanate (PEITC). Pharm Res 2003;20:1351-6.
- Yu X, Kensler T. Nrf2 as a target for cancer chemoprevention. Mutat Res 2005;591:93-102.
- Burton NC, Kensler TW, Guilarte TR. In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology 2006;27:1094-100.
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999;13:76-86.
- Dhakshinamoorthy S, Jaiswal AK. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene 2001;20:3906-17.
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 2003;23:8137-51.
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 2004; 24: 10 941–53.
- Velichkova M, Hasson T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell Biol 2005;25:4501-13.
- Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA 2004;101:2040-5.
- Li W, Yu SW, Kong AN. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J Biol Chem 2006; 281: 27 251–63.
- Dhakshinamoorthy S, Jaiswal AK. Small maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:quinone oxidoreductase1 gene. J Biol Chem 2000; 275: 40 134–41.
- Nair S, Xu C, Shen G, Hebbar V, Gopalakrishnan A, Hu R, et al. Pharmacogenomics of phenolic antioxidant butylated hydroxyanisole (BHA) in the small intestine and liver of Nrf2 knockout and C57BL/6J mice. Pharm Res 2006;23:2621-37.
- Jain AK, Bloom DA, Jaiswal AK. Nuclear import and export signals in control of Nrf2. J Biol Chem 2005; 280: 29 158–68.
- Bloom D, Dhakshinamoorthy S, Jaiswal AK. Site-directed mutagenesis of cysteine to serine in the DNA binding region of Nrf2 decreases its capacity to upregulate antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene 2002;21:2191-200.
- Shen G, Xu C, Hu R, Jain MR, Nair S, Lin W, et al. Comparison of (-)-epigallocatechin-3-gallate elicited liver and small intestine gene expression profiles between C57BL/6J mice and C57BL/6J/Nrf2 (-/-) mice. Pharm Res 2005;22:1805-20.
- Shen G, Xu C, Hu R, Jain MR, Gopalkrishnan A, Nair S, et al. Modulation of nuclear factor E2-related factor 2-mediated gene expression in mice liver and small intestine by cancer chemopreven-tive agent curcumin. Mol Cancer Ther 2006;5:39-51.
- Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, et al. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (-/-) mice. Cancer Lett 2006;243:170-92.
- Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, et al. Identification of Nrf2-regulated genes induced by chemopreven-tive isothiocyanate PEITC by oligonucleotide microarray. Life Sci 2006;79:1944-55.
- Nair S, Xu C, Shen G, Hebbar V, Gopalakrishnan A, Hu R, et al. Toxicogenomics of endoplasmic reticulum stress inducer tunica-mycin in the small intestine and liver of Nrf2 knockout and C57BL/6J Mice. Toxicol Lett 2006;168:21-39.
- Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001;6:857-68.
- Lin W, Shen G, Yuan X, Jain MR, Yu S, Zhang A, et al. Regulation of Nrf2 transactivation domain activity by p160 RAC3/SRC3 and other nuclear co-regulators. J Biochem Mol Biol 2006;39:304-10.
- Chen H, Tini M, Evans RM. HATs on and beyond chromatin. Curr Opin Cell Biol 2001;13:218-24.
- Ceribelli M, Alcalay M, Vigano MA, Mantovani R. Repression of new p53 targets revealed by ChIP on chip experiments. Cell Cycle 2006;5:1102-10.
- Talalay P. Chemoprotection against cancer by induction of phase 2 enzymes. Biofactors 2000;12:5-11.
- Zhang J, Ohta T, Maruyama A, Hosoya T, Nishikawa K, Maher JM, et al. BRG1 interacts with Nrf2 to selectively mediate HO-1 induction in response to oxidative stress. Mol Cell Biol 2006;26:7942-52.
- Wang W, Chan JY. Nrf1 is targeted to the endoplasmic reticulum membrane by an N-terminal transmembrane domain. Inhibition of nuclear translocation and transacting function. J Biol Chem 2006; 281: 19 676–87.
- Zhang K, Kaufman RJ. Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb Exp Pharmacol 2006.69-91.
- Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 2006;312:572-6.
- Srivastava RK, Sollott SJ, Khan L, Hansford R, Lakatta EG, Longo DL. Bcl-2 and Bcl-X(L) block thapsigargin-induced nitric oxide generation, c-Jun NH(2)-terminal kinase activity, and apoptosis. Mol Cell Biol 1999;19:5659-74.
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, et al. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994;369:156-60.
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000;287:664-6.
- Yu R, Jiao JJ, Duh JL, Tan TH, Kong AN. Phenethyl isothio-cyanate, a natural chemopreventive agent, activates c-Jun N-terminal kinase 1. Cancer Res 1996;56:2954-9.
- Garber K. Researchers target unfolded protein response in cancerous tumor growth. J Natl Cancer Inst 2006;98:512-4.
- Jones DP. Extracellular redox state: refining the definition of oxidative stress in aging. Rejuvenation Res 2006;9:169-81.
- Catarzi S, Biagioni C, Giannoni E, Favilli F, Marcucci T, Iantomasi T, . Redox regulation of platelet-derived-growth-factor-receptor: role of NADPH-oxidase and c-Src tyrosine kinase. Biochim Biophys Acta 2005; 1745: 166–75.
- Cheng YJ, Liu MY. Modulation of tumor necrosis factor-alpha and oxidative stress through protein kinase C and P42/44 mitogen-activated protein kinase in lead increases lipopolysaccharide-induced liver damage in rats. Shock 2005;24:188-93.
- Engers R, Springer E, Kehren V, Simic T, Young DA, Beier J, et al. Rac upregulates tissue inhibitor of metalloproteinase-1 expression by redox-dependent activation of extracellular signal-regulated kinase signaling. Febs J 2006;273:4754-69.
- Erker L, Schubert R, Yakushiji H, Barlow C, Larson D, Mitchell JB, et al. Cancer chemoprevention by the antioxidant tempol acts partially via the p53 tumor suppressor. Hum Mol Genet 2005;14:1699-708.
- Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006;10:241-52.
- Xu C, Shen G, Chen C, Gelinas C, Kong AN. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene 2005;24:4486-95.
- Shen HM, Pervaiz S. TNF receptor superfamily-induced cell death: redox-dependent execution. Faseb J 2006;20:1589-98.
- Han D, Hanawa N, Saberi B, Kaplowitz N. Hydrogen peroxide and redox modulation sensitize primary mouse hepatocytes to TNF-induced apoptosis. Free Radic Biol Med 2006;41:627-39.
- Arai RJ, Masutani H, Yodoi J, Debbas V, Laurindo FR, Stern A, et al. Nitric oxide induces thioredoxin-1 nuclear translocation: possible association with the p21Ras survival pathway. Biochem Biophys Res Commun 2006;348:1254-60.
- Shen G, Xu C, Chen C, Hebbar V, Kong AN. p53-independent G1 cell cycle arrest of human colon carcinoma cells HT-29 by sulforaphane is associated with induction of p21CIP1 and inhibition of expression of cyclin D1. Cancer Chemother Pharmacol 2006;57:317-27.
- Luo T, Xia Z. A small dose of hydrogen peroxide enhances tumor necrosis factor-alpha toxicity in inducing human vascular endothelial cell apoptosis: reversal with propofol. Anesth Analg 2006;103:110-6.
- Ge Y, Montano I, Rustici G, Freebern WJ, Haggerty CM, Cui W, et al. Selective leukemic cell killing by a novel functional class of thalidomide analogs. Blood 2006;108:4126-35.
- Macho A, Calzado MA, Munoz-Blanco J, Gomez-Diaz C, Gajate C, Mollinedo F, et al. Selective induction of apoptosis by capsaicin in transformed cells: the role of reactive oxygen species and calcium. Cell Death Differ 1999;6:155-65.
- Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulation of glutaredoxin-1 expression in a mouse model of allergic airway disease. Am J Respir Cell Mol Biol 2007;36:147-51.