
Antisense oligonucleotides targeting midkine inhibit tumor growth in an in situ human hepatocellular carcinoma model1
Introduction
Hepatocellular carcinoma (HCC), one of the most common malignant tumors worldwide, is often diagnosed at an advanced stage when most potentially curative therapies such as resection, transplantation, or percutaneous and transarterial interventions are of limited efficacy[1–4]. The fact that HCC is resistant to conventional chemotherapy, and is rarely amenable to radiotherapy, leaves this disease with no effective therapeutic options and poor prognosis[5–7]. Therefore, the development of more effective therapeutic tools and strategies is much needed. Despite the phenotypically and genetically heterogeneous tumors of HCC, recent insights into the biology of HCC suggest that certain signaling pathways and molecular alterations are likely to play essential roles in HCC development by promoting cell growth and survival. The identification of such mechanisms may open new avenues for the prevention and treatment of HCC through the development of targeted therapies.
Dysregulation of growth factors, receptors and their downstream signaling pathway components represent a central pro-tumorigenic principle in human hepatocarcinogenesis. In particular, the insulin-like growth factor/IGF-1 receptor (IGF/IGF-1R), hepatocyte growth factor (HGF)[8], Wnt[9], transforming growth factor alpha/epidermal growth factor receptor (TGF-α/EGFR)[10], and TGF-β/TGF-βR[11,12] pathways contribute to proliferation, anti-apoptosis, and invasive behavior of tumor cells. Midkine (MK) was first identified in embryonic carcinoma cells in early stages during retinoic acid-induced differentiation[13]. It has been reported that MK is overexpressed in HCC[14–16], whereas in normal adult tissues, MK is low or undetectable[17,18]. MK promotes the survival[19,20], growth[21,22], and migration[23–25] of many cells, which in combination may contribute to oncogenesis and tumor progression. Therefore, MK may be a promising target for cancer therapy. In fact, several studies have been reported that antisense targeting MK inhibits tumor growth, such as human prostate cells, colon carcinoma cells[26], and mouse rectal carcinoma cells[27].
We previously confirmed that MK-AS could significantly inhibit growth of hepatocellular cells including HepG2, SMMC-7721, and BEL-7402[28]. The aim of this study was to evaluate the in vivo antitumor effects of MK-AS in an in situ human hepatocellular carcinoma model in mice.
Materials and methods
Antisense oligodeoxynucleotides and drugs Antisense phosphorothioate oligonucleotide MK-AS (5'-CCCCGGGC-CGCCCTTCTTCA-3') targeting against 108 to 127 base position of MK mRNA and MK-Sen phosphorothioate oligonucleotide (5'-TGAAGAAGGGCGGCCCGGGG-3') were synthesized by an Applied Biosystems Model 391 DNA synthesizer on solid supports using Oligo Pilot II DNA (Amersham, Piscataway, NJ, USA) and purified by HPLC, (Waters Delta Prep 4000, Milford, MA, USA) with SOURCE 15Q (Amer-sham). The purity of the oligonucleotides was over 95%. The sense sequence was used as a control. 5-fluorouracil (5-Fu), purchased from the Shanghai Donghaipu Pharmaceuticals Company (Shanghai, China), was used as a positive control.
Animals and in situ human HCC model The virgin female Balb/c mice used in this experiments were obtained from the Academy of Military Medical Science (Beijing, China). All animal experiments were carried out according to the standards of animal care as outlined in the NIH guide for the Care and Use of Laboratory Animals. The human HCC tumor model was described previously[29]. Briefly, the HCM-Y89 tumor derived from a surgical specimen of HCC was cut into 1 mm×1 mm×1 mm tissues, and implanted into the liver of mice. Twenty days later, the mice treated with or without drugs were all killed. The tumors were removed and fixed in neutral buffered 10% formalin, processed by standard methods, embedded in paraffin, sectioned and stained with hematoxylin-eosin (HE). It is important to note that the HCC model maintains various features similar with clinical liver cancer patients including local growth, regional invasion, lymph nodes and pulmonary metastasis, peritoneal seeding with bloody ascites, and secretion of alpha-fetoprotein (AFP) in the recipient animals[29].
Treatment of in situ HCC xenograft with MK-AS and anticancer drugs Two days after the in situ HCC models were established, the tail veins of the mice were injected with saline (vehicle control), MK-AS (25, 50, and 100 mg·kg–1·d–1) or (5-Fu10 mg·kg–1·d–1) everyday for 20 d. Each group had 8 mice. The body weight and general physical status of the animal were recorded daily. At the endpoint of the study, the mice were killed by cervical dislocation and the tumors were removed and weighed. The tumor sizes were monitored with calipers; the tumor volume (V, mm3) was calculated as (L×W2)/2, where L=length (mm) and W=width (mm). The percentage of tumor growth inhibition was calculated as: Inhibitory rate (%)=(Wcontrol-Wtreat)/Wcontrol×100.
Histopathological analysis The tumor tissues were excised and fixed in 10% buffered formalin. Representative fragments were embedded in paraffin, and stained with HE for microscopic observations.
Detection of plasma AFP concentration Animal serum was collected after the mice were killed. Then, the plasma AFP concentrations were detected by radioimmunoassay (RIA) according to the manufacturer’s instructions.
Western blot analysis The tumor tissues were lysed with lysis buffer [50 mmol/L Tris-HCl (pH 7.4), 0.5 mmol/L EDTA, 0.5% NP40, and 150 mmol/L NaCl] in the presence of protease inhibitors. The lysates were centrifuged at 15 000 ×g for 15 min to remove debris. Proteins sample (30 µg) were separated by 12% SDS-PAGE gel and transferred onto polyvinylidene difluoride (PVDF) membranes (Hybond-polyvinylidene difluoride membranes, Amersham). p53, Bax, Bcl-2, caspase-3 and the MK protein were identified using the relative primary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The reactive band was visualized with an ECL-plus Detection Kit (Amersham) and scanned by Gel Doc 1000 (Bio-Rad, Hercules, CA, USA). β-actin was used as a control.
Statistical analysis Data were expressed as mean±SD. Statistical analysis was carried out using Student’s t-test (two-tailed); P<0.05 indicated statistical significance.
Results
Effects of MK-AS treatment on in situ HCC xenograft growth In the present study, we used an in situ mouse HCC model to evaluate the antitumor activity of MK-AS. In order to exclude the nonspecific effect of oligonucleotides, sense sequence (MK-Sen) was used as a control. After establishing the model for 2 d, MK-AS with different doses (25, 50, and 100 mg·kg–1·d–1), MK-Sen with 50 mg·kg–1·d–1, 5-Fu with 10 mg·kg–1·d–1, and saline were administered through the tail vein for 20 d. The tumors were removed after the mice were killed. The tumors were then measured and weighed. Table 1 shows the final tumor volumes after a 20 d treatment. Results showed that the tumor volumes decreased in the MK-AS treatment group compared with the saline control group (P<0.01). However, for the sense oligonucleotide treatment group, there was also a clear decrease in tumor volume compared with the saline group (P<0.05). No inflammatory infiltrate was observed surrounding the solid tumor (data not shown). In addition, MK-AS treatment also resulted in a significant inhibition of tumor weight compared to the sense or saline-treated mice (Table 2). The highest inhibitory efficacy was a 65.89% contrast to the saline group, but for the sense oligonucleotide, the inhibitory rate was only a 19.38% contrast to the saline group. The inhibitory rate of 50 mg·kg–1·d–1 was 50.39% compared to the sense treatment group (P<0.01). The effect of MK-AS on the inhibition of tumor proliferation was dose-dependent (Table 2). The mean body weight of the mice was not significantly different between the groups from the beginning to the end (data not shown). It is reported that a series of guanosine-rich, phosphodiester oligodeoxynucleotides can strongly inhibit proliferation in a number of human tumor cell lines[30]. On the basis, in this experiment, we thought that the induced decline of tumor volume and weight by MK-Sen might be related to its guanosine-rich, which is due to its binding activity with some nucleolin.
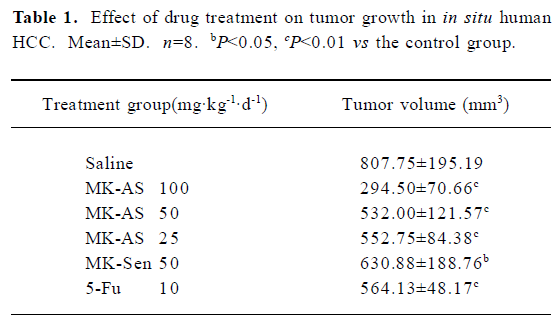
Full table
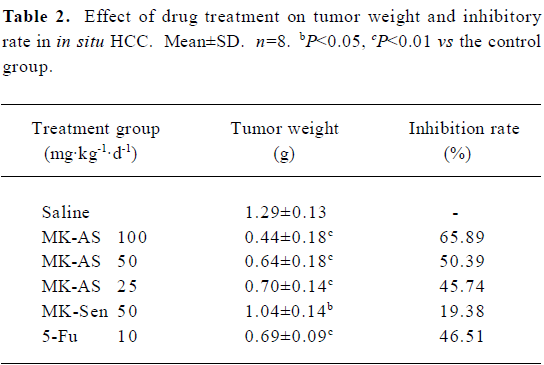
Full table
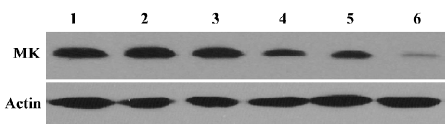
Effect of MK-AS treatment on MK expression in in situ hepatocellular carcinoma xenografts Our previous studies showed antisense oligonucleotides targeting against MK downregulated MK expression in hepatocellular carcinoma cells. Then, we measured the effect of MK-AS on MK expression in in situ hepatocellular carcinoma xenografts. The results indicated that the MK-AS efficiently decreased MK protein content in a dose-dependent manner (Figure 1). At the same time, we also observed that 5-Fu showed no effect on MK expression.
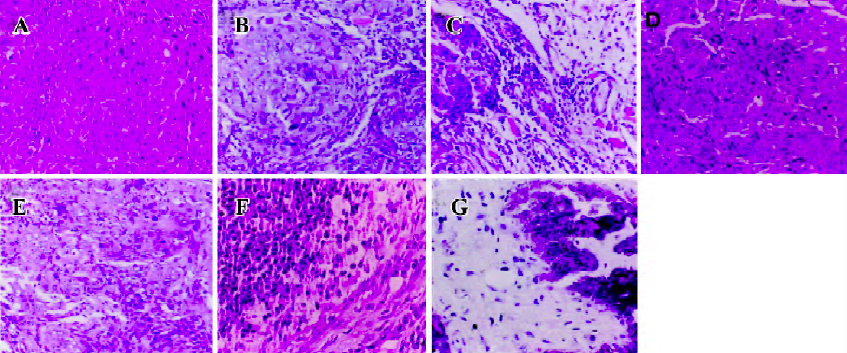
Histopathological analysis The morphology of tumors from MK-AS, MK-Sen, 5-Fu, and the saline-treated mice were evaluated by HE staining of the histological sections. The tumors were excised at the endpoint of treatment of each protocol. Figure 2 shows the representative sections of tumors from each experimental group. The tumors from the mice treated with MK-AS or 5-Fu showed a marked increase in the necrotic area compared with the saline-treated or normal animals, which suggested that MK-AS treatment induced HCC necrosis in vivo.
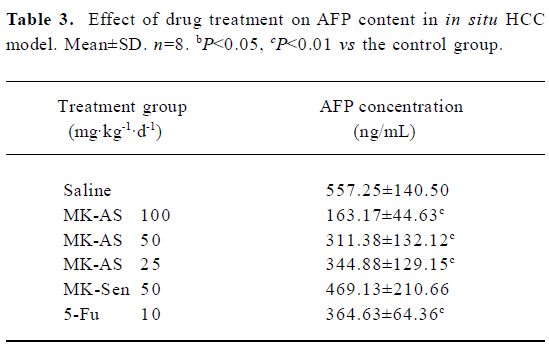
Inhibition of plasma AFP secretion with MK-AS treatment AFP is often expressed highly in fetal liver, the gastrointestinal tract, and the yolk sack, but it is transcriptionally downregulated after birth, and frequently re-expressed in HCC. Therefore, it is often used as an indicator of HCC[29]. In this experiment, we used RIA to detect serum AFP concentration at the endpoint of treatment. Table 3 shows that MK-AS significantly decreased AFP secretion at 100, 50, and 25 mg·kg-1·d-1 compared with the saline group, indicating that there were fewer liver tumor cells in the MK-AS treated mice and reduced circulating AFP.
Full table
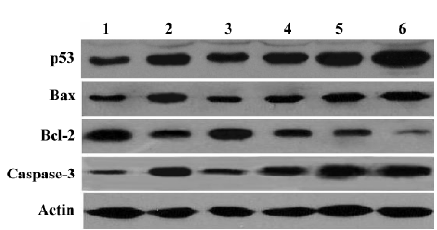
Effect of MK-AS on apoptosis-related protein expression Recently, apoptosis has been implicated as one of the end points of cells exposed to chemotherapeutic agents. p53, Bax, and Bcl-2 are involved in chemotherapy-induced apoptosis, but in a cell type-dependent manner[31–34]. In this experiment, we also measured the effect of MK-AS on these proteins’ expression. Interestingly, we observed that Bax, caspase-3, and p53 were upregulated in the MK-AS- and 5-Fu-treated groups, however, Bcl-2 decreased. These data suggested that modulation of pro-apoptotic and apoptotic protein expression was an important mechanism for MK-AS inhibiting tumor growth.
Discussion
In the present study, we first analyzed the effect of MK-AS administration on MK protein content in an in situ human hepatocellular carcinoma model. As measured by Western blotting, the MK-AS compounds efficiently down-regulated the MK expression level in a dose-dependent manner. However, MK-Sen and 5-Fu showed no effect on MK expression (Figure 1). Then, we determined the systemic administration of an in situ mouse HCC model. The results indicated that MK-AS administration resulted in a significant inhibition of the in situ mouse HCC model. Additionally, the histopathological results indicated that MK-AS could induce tumor necrosis. It needs to be noted that the experimental model of HCC has some advantages compared with the inoculated tumor model[29]. The tumor line originates from human HCC. It maintains the complete characteristics of human HCC tissue, such as AFP secretion and drug sensitivity. In addition, the pathological evidence also suggests that this model exhibits various features seen in clinical HCC patients. Therefore, the information provided by this model can reflect the true clinical results of patients in some ways. AFP is a secretary protein that is heterogeneously glycosylated.
AFP is usually expressed at high concentrations as mentioned earlier. It is transcriptionally downregulated after birth and frequently re-expressed in HCC. Therefore, it is always used as a diagnostic marker for tumors. Then we analyzed the drug treatment of AFP content in the serum. The results showed that serum AFP secretion was significantly inhibited in a dose-dependent manner in the MK-AS or 5-Fu treatment groups compared with the saline group. This result provided evidence that MK-AS could restrain HCC in vivo.
Up until now, there have been several reports about the mechanisms of pro-tumorigenesis by MK. It is reported that MK activates mitogen-activated protein kinase (MAPK) pathways and promotes cell growth[35]. Additionally, MK also activates extracellular signal-regulated kinases 1 and 2, which are well known as signal transducers acting downstream several receptors[36]. The activated MAPK pathway was thought to downregulate caspase-3 activity in neurons[35]. It was also believed that MK could induce phosphorylation of protein kinase B (AKT, Ser473 and Thr308), which promotes a series of anti-apoptosis pathways in cells. Interest-ingly, we also observed that MK-AS modulated expression of several proteins including Bax, Bcl-2, p53, and caspase-3 in vivo (Figure 3). We observed that caspase-3 protein content increased in MK-AS treated groups (Figure 3). The p53 tumor-suppressor gene is involved in cell growth control, arrest, and apoptosis. It was reported that, whatever p53 status, 5-Fu altered p53 transcriptional and translational regulation leading to the upregulation of the p53 protein. Moreover, after 5-Fu exposure, Bax and Bcl-2 protein regulation was under p53 protein control, and Bcl-2 or Bax induction and the Bcl-2/Bax protein ratio correlated to 5-Fu sensitivity[34]. In our experiments, we also observed these similar results. Furthermore, we observed that MK-AS administration increased p53, Bax, and caspase-3 protein content and decreased Bcl-2 content. This suggest that MK-AS has some similar functions with 5-Fu to restrain HCC by inducing some pro-apoptosis-related protein expression and inhibiting anti-apoptotic protein expression. It is well accepted that there is a significant correlation between chemosensitivity and Bcl-2 to the Bax ratio[34]. However, MK-AS administration can decrease the ratio in our experiment, so MK-AS can be used in combination therapy on human tumors.
In summary, we have for the first time directly addressed the potential therapeutic role of MK-AS in an in situ human HCC model. Significant inhibition of HCC growth is achieved by MK-AS, indicating that MK-AS has the possibility to be developed as an effective antitumor agent.
References
- Okuda K. Hepatocellular carcinoma. J Hepatol 2000;32:225-37.
- Befeler AS, Di Bisceglie AM. Hepatocellular carcinoma: diagnosis and treatment. Gastroenterology 2002;122:1609-161.
- Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet 2003;362:1907-17.
- Marrero JA. Hepatocellular carcinoma. Curr Opin Gastroenterol 2006;22:248-53.
- Mineura M, Tanimura H, Tabor E. Overexpression of multidrug resistance genes MDR1 and cMOAT in human hepatocellular carcinoma and hepatoblastoma cell lines. Int J Oncol 1999;15:559-63.
- Huang M, Liu G. The study of innate drug resistance of human hepatocellular carcinoma Bel7402 cell line. Cancer Lett 1999;135:97-105.
- Nies AT, Konig J, Pfannschimdt M, Klar E, Hofmann WJ, Keppler D. Expression of the multidrug resistance proteins MRP2 and MRP3 in human hepatocellular carcinoma. Int J Cancer 2001;94:492-9.
- Heideman DA, Overmeer RM, van Beusechem VW, Lamers WH, Hakvoort TB, Snijders PJ, et al. Inhibition of angiogenesis and HGF-cMET-elicited malignant processes in human hepatocellular carcinoma cells using adenoviral vector-mediated NK4 gene therapy. Cancer Gene Ther 2005;12:954-62.
- Lee HC, Kim M, Wands JR. Wnt/frizzled signaling in hepatocellular carcinoma. Front Biosci 2006;11:1901-15.
- Han C, Michalopoulos GK, Wu T. Prostaglandin E2 receptor EP1 transactivates EGFR/MET receptor tyrosine kinases and enhances invasiveness in human hepatocellular carcinoma cells. J Cell Physiol 2006;207:261-70.
- Ikeguchi M, Iwamoto A, Taniguchi K, Katano K, Hirooka Y. The gene expression level of transforming growth factor-beta (TGF-beta) as a biological prognostic marker of hepatocellular carcinoma. J Exp Clin Cancer Res 2005;24:415-21.
- Musch A, Rabe C, Paik MD, Berna MJ, Schmitz V, Hoffmann P, et al. Altered expression of TGF-beta receptors in hepatocellular carcinoma—effects of a constitutively active TGF-beta type I receptor mutant. Digestion 2005;71:78-91.
- Kadomatsu K, Tomomura M, Muramatsu T. cDNA cloning and sequencing of a new gene intensely expressed in early differentiation stages of embryonal carcinoma cells and in mid-gestation period of mouse embryogenesis. Biochem Biophys Res Commun 1988;151:1312-8.
- Tomizawa M, Yu L, Wada A, Tamaoki T, Kadomatsu K, Muramatsu T, et al. A promoter region of the midkine gene that is frequently expressed in human hepatocellular carcinoma can activate a suicide gene as effectively as the alpha-fetoprotein promoter. Br J Cancer 2003;89:1086-90.
- Yin Z, Luo X, Kang X, Wu Z, Qian H, Wu M. Correlation between midkine protein overexpression and intrahepatic metastasis in hepatocellular carcinoma. Zhonghua Zhong Liu Za Zhi 2002;24:27-9. Chinese..
- Kato M, Shinozawa T, Kato S, Awaya A, Terada T. Increased midkine expression in hepatocellular carcinoma. Arch Pathol Lab Med 2000;124:848-52.
- Li YS, Milner PG, Chauhan AK, Watson MA, Hoffman RM, Kodner CM, et al. Cloning and expression of a developmentally regulated protein that induces mitogenic and neurite outgrowth activity. Science 1990;250:1690-4.
- Tsutsui J, Kadomatsu K, Matsubara S, Nakagawa A, Hamanoue M, Takao S, et al. A new family of heparin-binding growth differentiation factors: increased midkine expression in Wilms’ tumor and other human carcinomas. Cancer Res 1993;53:1281-5.
- Owada K, Sanjo N, Kobayashi T, Mizusawa H, Muramatsu H, Muramatsu T, et al. Midkine inhibits caspase-dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. J Neurochem 1999;73:2084-92.
- Qi M, Ikematsu S, Ichihara-Tanaka K, Sakuma S, Muramatsu T, Kadomatsu K. Midkine rescues Wilms’ tumor cells from cisplatin-induced apoptosis: regulation of Bcl-2 expression by midkine. J Biochem 2000;127:269-77.
- Muramatsu H, Muramatsu T. Purification of recombinant midkine and examination of its biological activities: functional comparison of new heparin binding factors. Biochem Biophys Res Commun 1991;177:652-8.
- Muramatsu H, Shirahama H, Yonezawa S, Maruta H, Muramatsu T. Midkine (MK), a retinoic acid-inducible growth/differentiation factor: immunochemical evidence for the function and distribution. Dev Biol 1993;159:392-402.
- Takada T, Toriyama K, Muramatsu H, Song XJ, Torii S, Muramatsu T. Midkine, a retinoic acid-inducible heparin-binding cytokine in inflammatory responses: chemotactic activity to neutrophils and association with inflammatory synovitis. J Biochem 1997;122:453-8.
- Maeda N, Ichihara-Tanaka K, Kimura T, Kadomatsu K, Muramatsu T, Noda M. A receptor-like protein-tyrosine phosphatase PTPzeta/RPTPbeta binds a heparin-binding growth factor midkine. Involvement of arginine 78 of midkine in the high affinity binding to PTPzeta. J Biol Chem 1999; 274: 12 474–9.
- Horiba M, Kadomatsu K, Nakamura E, Muramatsu H, Ikematsu S, Sakuma S, et al. Neointima formation in a restenosis model is suppressed in midkine-deficient mice. J Clin Invest 2000;105:489-95.
- Takei Y, Kadomatsu K, Yuasa K, Sato W, Muramatsu T. Morpholino antisense oligomer targeting human midkine: its application for cancer therapy. Int J Cancer 2005;114:490-7.
- Takei Y, Kadomatsu K, Matsuo S, Itoh H, Nakazawa K, Kubota S, et al. Antisense oligodeoxynucleotide targeted to midkine, a heparin-binding growth factor, suppresses tumorigenicity of mouse rectal carcinoma cells. Cancer Res 2001;61:8486-91.
- Dai LC, Wang X, Yao X, Lu YL, Ping JL, He JF. Antisense oligonucleotides targeting midkine induced apoptosis and increased chemosensitivity in hepatocellular carcinoma cells. Acta Pharmacol Sin 2006;27:1630-6.
- Lin RX, Tuo CW, Lv QJ, Zhang W, Wang SQ. Inhibition of tumor growth and metastasis with antisense oligonucleotides (Cantide) targeting hTERT in an in situ human hepatocellular carcinoma model.. Acta Pharmacol Sin 2005;26:762-8.
- Bates PJ, Kahlon JB, Thomas SD, Trent JO, Miller DM. Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J Biol Chem 1999; 274: 26 369–77.
- Nita ME, Nagawa H, Tominaga O, Tsuno N, Fujii S, Sasaki S, et al. 5-Fluorouracil induces apoptosis in human colon cancer cell lines with modulation of Bcl-2 family proteins. Br J Cancer 1998;78:986-92.
- Kanda T, Yokosuka O, Imazeki F, Arai M, Saisho H. Enhanced sensitivity of human hepatoma cells to 5-fluorouracil by small interfering RNA targeting Bcl-2. DNA Cell Biol 2005;24:805-9.
- Sasaki M, Kumazaki T, Tanimoto K, Nishivama M. Bcl-2 in cancer and normal tissue cells as a prediction marker of response to 5-fluorouracil. Int J Oncol 2003;22:181-6.
- Mirjolet JF, Barberi-Heyob M, Didelot C, Peyrat JP, Abecassis J, Millon R, et al. Bcl-2/Bax protein ratio predicts 5-fluorouracil sensitivity independently of p53 status. Br J Cancer 2000;83:1380-6.
- Ohuchida T, Okamoto K, Akahane K, Higure A, Todoroki H, Abe Y, et al. Midkine protects hepatocellular carcinoma cells against TRAIL-mediated apoptosis through down-regulation of caspase-3 activity. Cancer 2004;100:2430-6.
- Owada K, Sanjo N, Kobayashi T, Mizusawa H, Muramatsu H, Muramatsu T, et al. Midkine inhibits caspase-dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. J Neurochem 1999;73:2084-92.