
Ceramide induces apoptosis in human lung adenocarcinoma A549 cells through mitogen-activated protein kinases
Introduction
Ceramide is a membrane sphingolipid that has recently emerged as a second messenger involved in the induction of apoptosis[1–4]. It can be generated either by sphingomye-linases or de novo synthesis. It plays an important role in activating cell death signals that are initiated by stress stimuli such as chemotherapeutic agents, cytokines, and ionizing radiation. Recent research indicates that exogenous ceramide causes apoptotic cell death in human lung adenocarcinoma A549 cell cultures[5–7].
The target molecules and terminal effectors of the ceramide-mediated apoptotic pathway have not been completely defined, and in lung cells they are unknown. Mitogen-activated protein kinases (MAPK) are a diverse superfamily of serine/threonine kinases that are conserved phylogenetically. There are 3 “classic” MAPK families: extracellular signal-regulated kinases (ERK1 and 2), the p38 MAPK (p38 α, β, δ, and γ), and c-Jun N-terminal kinases (JNK 1, 2, and 3)[8]. Individual MAPK is phosphorylated and activated by MEK (mitogen-activated protein kinase kinase, protein kinase that is usually specific for particular MAPK) and controls distinct cellular processes. Ceramide regulation of ERK1/2 activity is complex. There are certain cellular models where ceramide treatment may cause the activation of ERK1/2, whereas it appears to have an inhibitory effect in other models[9,10]. Under certain conditions, ceramide activates p38 and JNK MAPK in postmitotic cells, a step that may be critical for ceramide-induced apoptosis. Recent studies indicate that specific protease activation by ceramide may play a crucial role in the apoptotic process. These proteases, termed caspases for cysteinyl aspartate-specific protease, are members of a cysteine protease family. Studies with ceramide analogs or chemotherapeutic agents suggest that ceramide accumulation induces the activation of caspase-3[11,12].
Targeting the ceramide metabolic pathway is an attractive strategy to induce lung cancer cells to become apoptotic. However, a better understanding of the mechanisms leading to ceramide-mediated apoptosis is required. The role of the p38 MAPK in ceramide-induced apoptosis has been contro-versial, and a positive association has been identified, but mainly in neuronal cells[13,14]. On the other hand, although some reports have demonstrated that exogenous ceramide causes apoptotic cell death in human lung adenocarcinoma A549 cell cultures[5–7], the exact roles for MAPK in ceramide-induced cell death in A549 cells are unknown. Therefore, in order to provide experimental data for further research on the signal transduction of apoptosis in lung adenocarcinoma cells, we examined the effects of C2-ceramide (a non-natural, but cell-permeable analog of the endogenous long-chain ceramides) administration on several members of the MAPK superfamily and caspase-3 in A549 cells.
Materials and methods
Materials Cell culture growth media, buffers, and fetal bovine serum (FBS) were obtained from Life Technologies (Grand Island, NY, USA). C2-ceramide was from Sigma Chemical (St Louis, MO, USA). SB-203580 and U0126 were from Promega (Madison, WI, USA). Cell counting kit-8 (CCK-8) was from Wako Fine Chemicals (Osaka, Japan). Propidium iodide (PI) staining reagents for flow cytometry were purchased from Sigma Chemical (St Louis, USA). Bradford protein assay and Western analysis reagents and equipment were from Bio-Rad (Hercules, CA, USA). The following antibodies used for Western blotting were from Cell Signaling Technology (Beverly, MA, USA): phospho-MEK1/2 (Ser217/221) antibody, phospho-p44/42 MAPK (Thr202/Tyr204) antibody, phospho- SAPK (stress-activated protein kinase)/JNK (Thr183/Tyr185) antibody, phospho-p38 MAPK (Thr180/Tyr182) antibody, caspase-3 antibody, and cleaved caspase-3 antibody. p38 siRNA used siGenome SMARTselec-tion™ designed siRNA duplexes against p38 (MAPK14, Dharmacon, RNA technologies, Lafayette, CO, USA).
Cell culture Human lung adenocarcinoma A549 cells were obtained from the American Type Culture Collection (Rockville, MD, USA) and maintained in DMEM (Dulbecco’s Modified Eagle Medium) supplemented with 1% nonessential amino acids, 1% penicillin-streptomycin, and 10% FBS. Cells were seeded at a density of 1×106–5×106 cells/mL and incubated at 37 °C in a humidified atmosphere containing 5% CO2. After C2-ceramide (dissolved in DMSO) treatments, the cells were removed from the culture plates by incubation with 0.05% trypsin-EDTA (ethylenediaminetetraacetic acid) and washed twice with ice-cold phosphate buffered saline (PBS), and the cell pellets were used for the different assays.
Analysis of cell viability and apoptosis
Trypan blue exclusion assay The effects of C2-cera-mide on A549 cell growth were determined by trypan blue exclusion assay. In short, cells seeded as 3×105 cells/dish were treated in the absence or presence of various concentrations of C2-ceramide for 24 h. Then the cells were trypsinized and diluted in PBS. The floating dead cells in the medium and the cells that remained attached to the plates were then counted using a hematocytometer in the presence of trypan blue solution at a 1:1 ratio (v/v) (Sigma, USA) as described by the manufacturer.
Cell growth assay The measurement of cell proliferation was performed using 96-well cell culture plates (BD Falcon, Franklin Lakes, NJ, USA). A549 cells were seeded onto the plates at an initial density of approximately 0.1×105–0.5×105 cells in DMEM containing 10% FBS per well, and the total volume was 200 µL after adding different concentrations of C2-ceramide into each well. Where appropriate, cells were pretreated for 1 h with SB-203580 (2.5–20 µmol/L, a p38 MAPK inhibitor) or U0126 (1–6 µmol/L, a MEK inhibitor). Then the cells were incubated for 24 h to allow adhesion. Cells cultured without test material served as controls. The number of cells was determined using CCK-8. Ten μL of the CCK-8 solution was added to each well of the plate. The plate was incubated for 1 h in the humidified incubator (at 37 °C, 5% CO2). After incubation, cell growth was determined colorimetrically at 450 nm (Titertec Multiskan Plus, Flow Laboratory, McLean, VA, USA). The cell number was calculated from the absorbance value relative to a standard curve.
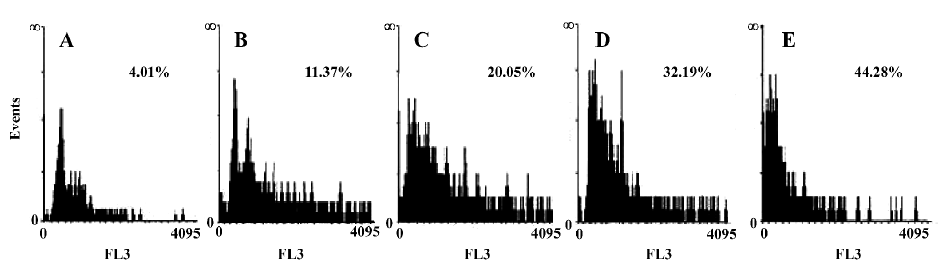
Cell apoptosis detected by fluorescence activated cell sorter (FACS) analysis After treatment with C2-ceramide(100 µmol/L) for 24 h, the cells were harvested by trypsinization and centrifuged at 400 ×g for 15 min, stained with PI (50 µg/mL), then immediately analyzed by flow cytometry (FACSCaliber, Becton Dickson, Franklin Lake, NJ, USA). Experiments were performed in triplicate and a total of 10 000 cells were analyzed in each individual experiment.
Western blotting Western blotting analysis was done as described previously[15]. In brief, cells were lysed in lysis buffer {1% Triton X, 5 mmol/L EDTA, 5 mmol/L EGTA [Ethyleneglycol bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid], and protease inhibitor cocktail (Sigma, USA) in PBS}, and the samples were centrifuged at 10 000 ×g for 10 min. Protein concentrations in the samples were determined with the Bradford protein assay kit (Bio-Rad, USA) and bovine serum albumin as the standard. Samples containing equal amounts of protein (40 µg)were resolved by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad, USA), blocked, and incubated overnight with specific primary antibodies in 0.1% Tween-20, 5% nonfat dried milk in 10 mmol/L Tris-HCl and 150 mmol/L NaCl (pH 7.5). Immunoreactive bands were visualized using the enhanced chemiluminescence Western blotting protocol (Amersham Pharmacia, Piscataway, New Jersey, USA).
p38 siRNA transfection Equally seeded wells of A549 cells were transiently transfected with 120 nmol/L siRNA using Lipofectamine Plus TM reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. As the control, cells were treated with Lipofectamine Plus TM reagent alone.
Statistical analysis All statistical analyses were performed by SPSS software. We used t-test. In all cases, the minimal requirement for establishing significance was 0.05.
Results
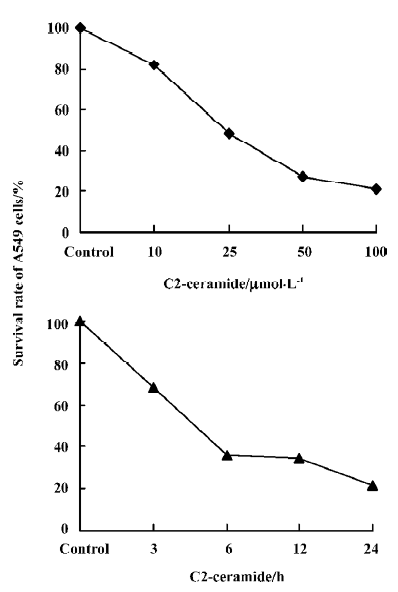
Detection of the effects of C2-ceramide on A549 cell survival and apoptosis The effects of C2-ceramide on A549 cells survival were examined by trypan blue exclusion assay, CCK-8 assay and FACS analysis. Treatment of A549 cells with increasing concentrations of C2-ceramide was done. At 24 h, the cells were harvested, and the survival rate of the A549 cells was measured and compared to time-matched controls. As shown in Figure 1, it was found that at the concentration of 10 µmol/L C2-ceramide, a 17.93% decrease in the cell survival rate was detected. Furthermore, in the presence of 50 µmol/L and 100 µmol/L C2-ceramide, substantial drop in the survival rate was observed, since 72.31% and 78.68% of cells, respectively, were killed. On the other hand, incubation with C2-ceramide (100 µmol/L) for 3, 6, 12, and 24 h resulted in the survival rate decreasing to 68.36%, 36.11%, 34.72%, and 21.32%, respectively (Figure 1). As demonstrated in Figure 2, the absorbance of C2-ceramide-treated cells decreased with the increasing concentration of C2-ceramide. Compared with the control (DMEM with A549 cells), at the concentration of 50 µmol/L C2-ceramide, the absorbance of A549 cells reduced to 85.26% (P<0.01). With concentrations of 100 and 200 µmol/L, there was a significant decrease in the absorbance of the A549 cells, which was 63.82% and 61.37%, respectively, versus the control (P<0.01). Through the dose-response experiments, we established that 100 µmol/L is one of the lowest concentrations of C2-ceramide capable of inducing a significant degree of apoptosis in A549 cells after 24 h of treatment. Therefore, this C2-ceramide concentration was used throughout our studies. As shown in Figure 3, C2-ceramide (100 µmol/L) induced apoptosis in A549 cells in a time-dependent fashion.

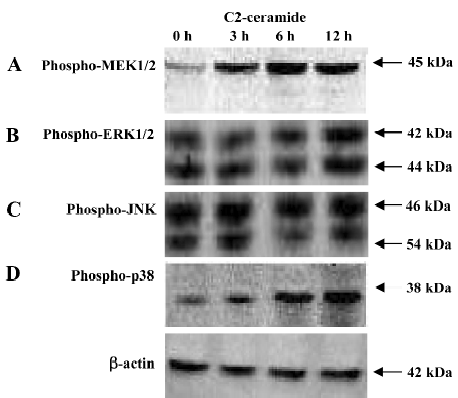
Changes of MAPK during C2-ceramide induced A549 cell death or apoptosis By Western blotting analysis, we examined the changes of MAPK during C2-ceramide induced A549 cell death or apoptosis. As demonstrated in Figure 4, C2-ceramide treatment of A549 cells caused major changes in the phosphorylation state of MAPK. Compared to the control samples (DMEM with A549 cells), in the C2-ceramide-treated A549 cells, the phosphorylation of MEK1/2 (Figure 4A) and p38 (Figure 4D) increased significantly in a time-dependent manner. These changes were evident as early as 3 h after C2-ceramide administration and were maximal between 6 and 12 h. But JNK (Figure 4B) and ERK1/2 (Figure 4C) phosphorylation showed no significant alteration during the same time frame.
Changes of caspase-3 during C2-ceramide induced A549 cell death or apoptosis The role of caspases in the execution phase of apoptosis is well established. The report that exposure of primary human lung cancer A549 airway epithelial cells to H2O2 induce a rapid accumulation of cellular ceramide and increase the apoptotic populations of these cells led us to examine the effects of C2-ceramide on caspase activation, focusing on caspase-3, the last caspase to be activated in the execution phase of apoptosis[2,16,17]. Compared with the control samples, in the C2-ceramide-treated A549 cells, procaspase-3 (an inactive form of caspase-3) increased remarkably as early as 6 h after C2-ceramide administration and was maximal at 12 h. The active form of caspase-3, cleaved caspase-3, was detected at 6 h and enhanced notably at 12 h after C2-ceramide treatment (Figure 5).

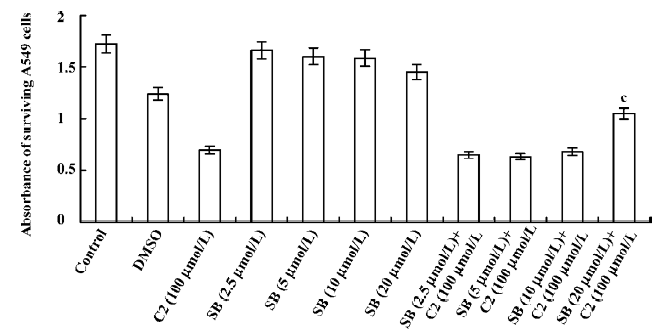
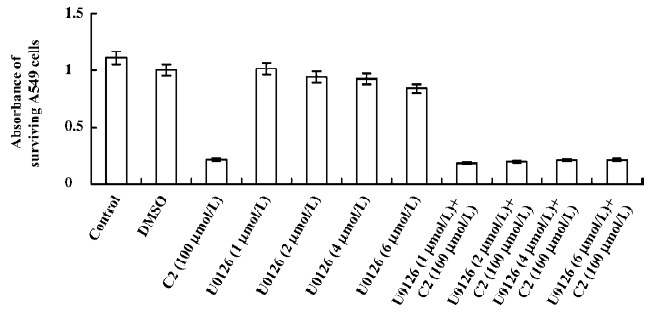
Effects of inhibitors of the p38 MAPK and MEK1/2 We used CCK-8 assay to determine the intervening effects of SB-203580 (pyridine imidazole compounds that specifically inhibit p38 MAPK α and β, but not p38 δ and γ) and U0126 (a specific inhibitor of the ERK1/2 activator kinases, MEK1/2). In contrast to the C2-ceramide-treated cells, SB-203580 pretreatment (at a concentration of 20 µmol/L) improved cell viability significantly after C2-ceramide treatment, whereas U0126 pretreatment had no significant effect on C2-ceramide induced cell death even at the concentration of 6 µmol/L (Figures 6, 7).
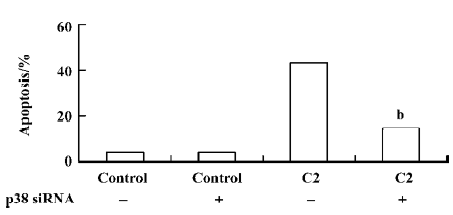
To investigate the vital role of the p38 MAPK in C2-ceramide induced apoptosis, we employed p38 siRNA. We confirmed our findings by transfecting the cells with p38 siRNA oligonucleotides and determining the rates of apoptosis following treatment with 100 µmol/L C2-ceramide. To determine the efficiency of p38 siRNA on inhibiting p38 expression, Western blotting was performed in the presence and absence of C2-ceramide. We found a marked decrease in p38 expression in the A549 cells following transfection with the siRNA at baseline and after treatment with 100 µmol/L C2-ceramide (Figure 8).We found that p38 siRNA had no effect on basal rates of apoptosis, but significantly decreased apoptosis induced by 100 µmol/L C2-ceramide compared to nontransfected cells (Figure 9). These data suggest that p38 activation mediates apoptosis induced by 100 µmol/L C2-ceramide in A549 cells.

Discussion
Recent experimental evidence supports a role for C2-ceramide (an exogenous analog of ceramide) in the induction and progression of human lung adenocarcinoma A549 cell death[5,6]. In the present report, we examine cell survival in A549 cells exposed to exogenous C2-ceramide and changes in MAPK phosphorylation and caspase-3 activation. C2-ceramide induced evident alterations in the phosphorylation of MEK1/2, and the p38 MAPK, but had no effect on ERK1/2 and JNK. The p38 inhibitor, but not the MEK1/2 inhibitor, partially attenuated ceramide-induced cell death. Our data also show C2-ceramide-induced apoptosis through the activation of caspase-3.
Cell cycle checkpoints are important mechanisms that ensure the proper execution of cell cycle events. MAPK are the major molecular players in cell cycle progression. Recently, many studies have shown that the inhibition of MAPK activity, which leads to cell cycle arrest, has turned out to be the productive strategy for the discovery and design of novel anticancer of cancer cells by causing cell cycle arrest and apoptosis. Therefore, we used specific inhibitors for the MEK1/2 and p38 MAPK pathways. The p38 MAPK inhibitor (SB-203580) partially attenuated ceramide-induced apoptosis, whereas the MEK1/2 inhibitor (U0126) had no significant effect on C2-ceramide-induced cell death. These results indicated that SB-203580, but not U0126, partially rescued cell death induced by C2-ceramide. Thus, the p38 MAPK was partially involved in C2-ceramide-induced A549 cell death or apoptosis. The high specificity demonstrated by these inhibitors for their enzyme targets, as well as the fact that we used moderate concentrations and included inactive controls, is consistent with our conclusion regarding the modulatory roles of MAPK in ceramide-induced apoptosis.
Although often associated with promoting cell survival, ERK1/2 activation has also been linked to cell death[9,10]. It appears that the ERK1/2 pathway can influence the balance between cell survival and cell death in both ways, depending of the specific cellular system and its condition. To date, the mechanisms underlying the deleterious effects of ERK1/2 remain unclear. In our experiments, although the phosphorylation of MEK1/2 was increased, C2-ceramide did not affect ERK1/2 in A549 cells. Moreover, U0126 did not attenu-ate A549 cell death significantly. This is partly because the MEK1/2 and ERK1/2 pathways were not crucial in the signaling cascades of C2-ceramide-induced A549 apoptosis.
Our results support and extend, but also differ in several significant regards, from those reported by another group. Although we confirmed a role for the p38 MAPK in ceramide-induced A549 cell death, Newton et al[7] found that ceramide was an efficient inducer of the ERK, but not the p38 MAPK. We believe that these dissimilarities reflect differences in ceramide treatment conditions and A549 culture systems. In our research, A549 cells were administrated C2-ceramide (100 µmol/L) for 3, 6, and 12 h, respectively. Newton et al only treated cells with C2-ceramide (100 µmol/L) for 15, 30, or 45 min, respectively. So the unobvious change of the p38 MAPK was probably due to the short duration of the ceramide treatment. Furthermore, the difference of the A549 culture system is also possibly responsible for the diversity of the results. We employed DMEM supplemented with 10% FBS in the cell culture system, whereas they used serum-free media in order to concentrate on observing the effects of ceramide on cyclooxygenase-2 prostaglandin E2 and nuclear factor-kappaB (a kind of intracellular signaling protein).
In our experiments, C2-ceramide did not influence the phosphorylation of JNK. This indicates that JNK did not participate in ceramide-induced apoptosis. Kurinna et al[5] reported that ceramide promoted apoptosis in A549 cells by a mechanism involving JNK. The JNK inhibitor SP 600125 proved effective at protecting cells from the lethal effects of ceramide. To understand which JNK-mediated pathway may be involved, a number of JNK target proteins were examined. At last they found that ceramide promoted the phosphorylation of Bim (a pro-apoptotic protein in Bcl-2 family members) and induced translocation of active JNK from the nucleus to the cytoplasm and mitochondrial fraction. Ceramide-mediated changes in the localization of JNK were consistent with the observed changes in the phosphorylation status of c-Jun and Bim. Furthermore, ceramide promoted Bim translocation to the mitochondria. The mitochondrial localization of Bim has been shown recently to promote apoptosis. These results suggest that JNK may participate in ceramide-induced apoptosis in A549 cells by a mechanism involving Bim. In conjunction with their results, JNK may play a role in ceramide-induced A549 cells apoptosis by other manners, but does not influence the activation of JNK.
Our results also show that C2-ceramide promoted the expressions of procaspase-3 and caspase-3 that is known to involve cell cycle arrest and apoptosis. Similarly, Ravid et al[5] elucidated the link between caspase-3 activation at the execution phase, and ceramide accumulation, at the commitment phase of apoptosis in A549 human lung adenocarcinoma cells. They concluded that ceramide accumulation precedes caspase-3 activation during apoptosis of A549 human lung adenocarcinoma cells. Caspases play critical roles in the initiation of apoptosis. It is well known that 2 apoptotic pathways called caspases dependent and independent are being reported in mammalian cells. Based on the substrate specificities and target proteins of caspases, caspases can be grouped as “apoptotic initiators”, such as caspase-8, and “apoptotic effector”, such as caspase-3. In our study, cleaved caspase-3 induced by C2-ceramide was observed at 6 h, and then up to 12 h (Figure 6). We also found that C2-ceramide decreased the expression of the pro-apoptotic proteins in Bcl-2 family members such as Bax and Bad in A549 cells. We also used flow cytometry assay for caspase-3 activity where it was indicated that C2-ceramide promoted the activity of caspase-3 (data not shown).
In summary, our results indicate that exogenous C2-ceramide induces apoptosis in human lung adenocarcinoma A549 cells. Several numbers of MAPK and caspase-3 were involved in the mechanisms of ceramide-induced apoptosis in A549 cells. Our research provides experimental data for further research on the signal transduction of apoptosis in lung adenocarcinoma cells. The further research on ceramide-induced apoptosis is a promising and encouraging career and will contribute to the exploration and development of new anticancer therapeutic drugs[18–23].
References
- Wang J, Hu XS, Shi JP. Sphingolipid and apoptosis. Sheng Li Ke Xue Jin Zhan 2003;34:217-21.
- Chan C, Goldkorn T. Ceramide path in human lung cell death. Am J Respir Cell Mol Biol 2000;22:460-8.
- Kolesnick R, Hannun YA. Ceramide and apoptosis. Trends Biochem Sci 1999;24:224-5.
- Perry DK. Ceramide and apoptosis. Biochem Soc Trans 1999;27:399-404.
- Kurinna SM, Tsao CC, Nica AF, Jiffar T, Ruvolo PP. Ceramide promotes apoptosis in lung cancer-derived A549 cells by a mechanism involving c-Jun NH2-terminal kinase. Cancer Res 2004;64:7852-6.
- Ravid T, Tsaba A, Gee P, Rasooly R, Medina EA, Goldkorn T. Ceramide accumulation precedes caspase-3 activation during apoptosis of A549 human lung adenocarcinoma cells. Am J Physiol Lung Cell Mol Physiol 2003;284:L1082-92.
- Newton R, Hart L, Chung KF, Barnes PJ. Ceramide induction of COX-2 and PGE(2) in pulmonary A549 cells does not involve activation of NF-kappaB. Biochem Biophys Res Commun 2000;277:675-9.
- Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol 2003;19:91-118.
- Shin CY, Lee YP, Lee TS, Song HJ, Sohn UD. C(2)-ceramide-induced circular smooth muscle cell contraction involves PKC-epsilon and p44/p42 MAPK activation in cat oesophagus. Mitogen-activated protein kinase. Cell Signal 2002;14:925-32.
- Willaime S, Vanhoutte P, Caboche J, Lemaigre-Dubreuil Y, Mariani J, Brugg B. Ceramide-induced apoptosis in cortical neurons is mediated by an increase in p38 phosphorylation and not by the decrease in ERK phosphorylation. Eur J Neurosci 2001;13:2037-46.
- Smyth MJ, Perry DK, Zhang J, Poirier GG, Hannun YA, Obeid LM. prICE: a downstream target for ceramide-induced apoptosis and for the inhibitory action of Bcl-2. Biochem J 1996;316:25-8.
- Sawada M, Nakashima S, Banno Y, Yamakawa H, Hayashi K, Takenaka K, et al. Ordering of ceramide formation, caspase activation, and Bax/ Bcl-2 expression during etoposide-induced apoptosis in C6 glioma cells. Cell Death Differ 2000;7:761-72.
- Stoica BA, Movsesyan VA, Knoblach SM, Faden AI. Ceramide induces neuronal apoptosis through mitogen-activated protein kinases and causes release of multiple mitochondrial proteins. Mol Cell Neurosci 2005;29:355-71.
- Willaime-Morawek S, Brami-Cherrier K, Mariani J, Caboche J, Brugg B. C-Jun N-terminal kinases/c-Jun and p38 pathways cooperate in ceramide-induced neuronal apoptosis. Neuroscience 2003;119:387-97.
- Ravid T, Avner R, Polak-Charcon S, Faust JR, Roitelman J. Impaired regulation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase degradation in lovastatin-resistant cells. J Biol Chem 1999;274:29341-51.
- Goldkorn T, Balaban N, Shannon M, Chea V, Matsukuma K, Gilchrist D, et al. H2O2 acts on cellular membranes to generate ceramide signaling and initiate apoptosis in tracheobronchial epithelial cells. J Cell Sci 1998;111:3209-20.
- Lavrentiadou SN, Chan C, Kawcak TN, Ravid T, Tsaba A, van der Vliet A, et al. Ceramide-mediated apoptosis in lung epithelial cells is regulated by glutathione. Am J Respir Cell Mol Biol 2001;25:676-84.
- Padron JM. Sphingolipids in anticancer therapy. Curr Med Chem 2006;13:755-70.
- Modrak DE, Gold DV, Goldenberg DM. Sphingolipid targets in cancer therapy. Mol Cancer Ther 2006;5:200-8.
- Siskind LJ. Mitochondrial ceramide and the induction of apoptosis. J Bioenerg Biomembr 2005;37:143-53.
- Sawai H, Domae N, Okazaki T. Current status and perspectives in ceramide-targeting molecular medicine. Curr Pharm Des 2005;11:2479-87.
- Jaffrezou JP, Laurent G. Ceramide: a new target in anticancer research? Bull Cancer 2004;91:E133-61.
- Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 2004;4:604-16.