
Involvement of hepatitis B X-interacting protein (HBXIP) in proliferation regulation of cells1
Introduction
The hepatitis B X-interacting protein (HBXIP), encoding a 9.6 kDa protein, was originally identified by its interaction with the C-terminus of the hepatitis B virus (HBV) X protein (HBX) and located at human chromosome 1p13.3[1]. It contained a putative leucine zipper motif and 2 consensus phosphorylation sites at threonines 12 and 36 for protein kinase C and casein kinase II. Investigation for the role of HBXIP in the HBV replication in hepatoma cells revealed that HBXIP reduced the replication of the wild-type HBV following transfection with a HBX-minus virus[1]. The transactivation effect of HBX on an activating protein-1 (AP-1) binding site as well as on the HBV enhancers was abolished by co-expression with the HBXIP. HBXIP could form complex with survivin, an anti-apoptotic protein that is overexpressed in most human cancers[2]. The complex of HBXIP and survivin binded pro-caspase-9 to prevent its recruitment to Apaf I, and thereby selectively suppressing apoptosis initiated via the mitochondrial/cytochrome c pathway. HBXIP was required for bipolar spindle formation and was a regulator of centrosome dynamics and cytokinesis in cells[3]. Recently, an interaction factor of HBXIP was identified, which was a human ATP-dependent RNA/DNA helicase hSuv3p. The nucleotide triphosphate (NTP)-dependent DNA/RNA DExH box helicase was predominantly localized in the mitochondria. It was found that the HBXIP-binding domain was important for mitochondrial import and the stability of the Suv3 protein[4]. Moreover, we found that HBXIP inhibited apoptosis induced by HBX in hepatoma cells[5]. We previously demonstrated that 8 types of the HBXIP gene were homologous (data not shown), such as (i) human fetal tissues of skeletal muscle, cardiac muscle and uterus muscle; (ii) human adult uterus muscle; (iii) mouse tissues of skeletal muscle, cardiac muscle and uterus muscle; and (iv) SM3 cells (a derivation of rabbit vascular smooth muscle), suggesting that HBXIP was a conserved protein in evolution.
Although some binding proteins of HBXIP have been identified in the last few years, the basic biological functions of HBXIP are still unclear. In the present study, we are interested in the basic biological functions of HBXIP and address whether HBXIP is able to influence cell proliferation by transfection with HBXIP gene in cells, such as breast cancer MCF-7 cells and hepatoma H7402 cells, and in normal liver L-O2 cells.
Materials and methods
Cancer cells and cell culture MCF-7 cells, H7402 cells[6], and L-O2 cells[6] were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 2 mmol/L glutamine and 100 U/mL penicillin, and 100 µg/mL streptomycin in humidified 5% CO2 at 37 oC.
Construction of the HBXIP gene in vectors Total RNA was extracted from H7402 cells using Trizol (Invitrogen, Carlsbad, CA, USA) reagent following the manufacturer’s recommendations. Prior to the first cDNA strand synthesis, total RNA was digested with RNase-free DNase I (TaKaRa, Tokyo, Japan) at 37 oC for 20 min and inactivated at 60 oC for 10 min. With 2 µg of total RNA as the template and oligo (dT) as the primers, the first cDNA strand was synthesized in a 20 μL reaction system with M-MLV reverse transcriptase (TaKaRa). One µL of cDNA template was used in a 50 µL reaction volume with rTaq DNA polymerase (TaKaRa) and oligonucleotide primers as follows: 5'-GACGAATTCATGG-AGGCGACCTTGGAGCA-3' (forward) and 5'-GATCTC-GAGTCAAGAGGCCATTTTGTGCA-3' (reverse). The resultant cDNA fragments were ligated into pET30a vector (termed pET30a-hbxip) and pGEX-4T-1 encoding glutathione S-transferase (GST) vector (termed pGEX-4T-hbxip), respec-tively. The pET30a-hbxip plasmids and pGEX-4T-hbxip plasmids were induced by 0.4 mmol/L isopropyl β-D-thiogalacto-pyranoside (IPTG) in Escherichia coli BL21 (DE3) at 37 oC for 4 h. The purified fusion protein of GST-HBXIP (35.6 kDa) expressed by pGEX-4T-hbxip plasmids was used to identify the specificity of rabbit anti-HBXIP antibody as the antigen.
Generation of antibody of rabbit anti-HBXIP pET30a-hbxip was induced by 0.4 mmol/L IPTG in Escherichia coli BL21 (DE3) at 37 oC for 4 h, and the HBXIP band was collected from the gel after SDS-PAGE electrophoresis, which was used as the immunogen. Equal amounts of antigen solution and adjuvant were mixed thoroughly. HBXIP antigen (0.1 mg) was administered by an intradermal injection of rabbit antibody. First immunizations were done with complete Freund’s adjuvant (Sigma, St Louis, MO, USA) and subsequent immunizations were done employing incomplete Freund’s adjuvant (Sigma) weekly for 5 times. The serum was harvested by bleeding at the sixth week. The rabbit antibody against HBXIP was purified from the immune sera by affinity purification.
Construction of RNA interference (RNAi) targeting the HBXIP gene in the vector According to a report[2], a synthesized 63 mer oligonucleotide containing a specific sequence for a targeting region of the HBXIP open reading frame was inserted into the pSilencer-3.0-H1 of the RNAi vector. The sequence of oligonucleotide 1 was as follows: 5'-GATCCGC- AGCTAAGCTAACCTCTGTTCAAGAGACAGAGGTTAG-CTTAGCTGCTTTTTTGGAAA-3'; the sequence of oligonucleotide 2 was as follows: 5'-AGCTTTTCCAAAAAAG-CAGCTAAGCTAACCTCTGTCTCTTGAACAGAGGTTA-GCTTAGCTGCG-3'. The 2 annealed complementary oligonucleotides were inserted into the BamH I/Hind III site of the pSilencer 3.0 H1 vector. After PCR and enzyme digestion, identification of the RNAi fragment of HBXIP inserted in the vector was performed by sequencing.
Transfection One day before transfection, MCF-7 cells, H7402 cells, and L-O2 cells were collected, and seeded into 6-well plates at 1×105 cells per well (n=3, each group). MCF-7, H7402, or L-O2 cells were transfected with 2 μg plasmids such as pcDNA3 empty vector, pcDNA3-hbxip encoding HBXIP[5], and pSilencer-hbxip respectively, using Lipofecta-mine 2000 (Invitrogen) according to the manufacturer’s instruction. The transfection mixture was removed after 6 h. Transfection efficiency in the cells was monitored by co-transfection of 0.2 µg pEGFP-C2 plasmid, which expresses green fluorescence protein (GFP). After 24 h transfection, 5-bromo-2-deoxyuridine (BrdU) incorporation assay was performed, and after 48 h transfection, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay, flow cytometry analysis, and Western blot analysis were followed to detect cell proliferation, cell cycle, and expression of proteins associated with proliferation.
MTT assay Cell proliferation was measured by MTT assay[7]. Briefly, an amount of 200 µL cell suspensions (5×103 cells/mL) was added to each well of 96-well plates and incubated at 37 oC for 48 h. After 48 h transfection (as described earlier), an amount of 20 µL MTT (5 mg/mL, Genview, Houston, TX, USA) was added and incubated at 37 oC for 4 h. After removing the supernatant, 200 µL dimethyl sulfoxide was added to resolve formazan crystals, and the value of the optical density was detected at 570 nm. The results are based on the cleavage of the tetrazolium salt by viable cells that were proportional to the number of living cells in the wells.
BrdU labeling and immunofluorescent staining The detailed procedures were followed accordingly[8]. In brief, the cells were seeded into 6-well plates and were grown overnight prior to transfection. All groups (n=3 in every group) were incubated with fresh medium containing 10 µmol/L BrdU (Sigma) for 4 h prior to immunofluorescence staining with mouse anti-BrdU antibody. The cells were fixed for 15 min with 4% paraformaldehyde in phosphate buffered saline (PBS). After 1 h incubation with PBS containing 2 mol/L HCl to denature DNA, cover slips were washed 3 times with 0.5% bovine serum albumin (BSA) and 0.5% Tween 20 in PBS, and incubated overnight (4 oC) with a mouse anti-BrdU antibody (NeoMarkers, Fremont, CA, USA) at 1:300 dilution. Reactions were developed using fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (Dako, Glostrup, Denmark) at 1:100 dilution for BrdU staining. The BrdU labeling index was assessed by point counting through a Nikon TE200 inverted microscope (Nikon, Tokyo, Japan) using a 40×objective lens. A total of 700–800 nuclei were counted in 6–8 representative fields. The labeling index was expressed as the number of positively-labeled nuclei/total number of nuclei. Propidium iodine (PI) (Sigma) staining for nuclei in 50 µg/mL was used as the control to all cells in each group.
Flow cytometry analysis After 48 h transfection as earlier described, the cells (1×106) were harvested by trypsinization and washed twice with PBS. Washed cells were resuspended in 0.6 mL PBS (pH 7.4), and fixed by the addition of 1.4 mL 100% ethanol at 4 oC overnight. The fixed cells were rinsed twice with PBS, and resuspended in propidium iodine (PI) solution, including 50 µg/mL PI and 50 µg/mL RNaseA (Sigma) in PBS without calcium and magnesium, and incubated at 37 oC for 30 min in the dark. Stained cells were passed through a nylon-mesh sieve to remove cell clumps and analyzed by a FACScan flow cytometer and Cell Quest analysis software (Becton Dickinson, San Jose, CA, USA). Flow cytometry analysis was repeated 3 times.
Western blot analysis For the identification of the generated rabbit anti-HBXIP antibody, cell lysates from MCF-7 and purified GST-HBXIP from BL21 (DE3) cells were examined by Western blot analysis. After washing twice in cold PBS, the cells were lysed with ice-cold lysis buffer (150 mmol/L NaCl, 20 mmol/L Tris-HCl, pH 7.4, 0.1% SDS, 1.0% Nonidet P-40, 0.5% Na-deoxycholate, 0.2 mmol/L phenylme-thylsulfonyl fluoride, and protein inhibitor cocktails). Lysates were centrifuged at 12 000×g for 20 min, and the supernatants were used as total cell lysates. The protein concentration was determined by Bradford protein assay (Bio-Rad, Hercules, CA, USA). A quantity of 30 µg total protein per lane was separated by SDS-PAGE and transferred onto polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). Membranes were blocked with 5.0% milk powder in 0.05% Tween-PBS, incubated with the specific antibodies such as rabbit anti-HBXIP (1:1000 dilution) or anti-GST antibody (Tiangen, Beijing, China, 1:800 dilution), at room temperature for 2 h, followed by a peroxidase-conjugated secondary antibody diluted in 0.3% BSA/Tween-PBS at room temperature for 1 h. Detection of the target proteins on the membranes was performed using the enhanced chemiluminescence (ECL) system Western Blotting Detection Reagents (Amersham Biosciences, Buckinghamshire, UK). All experiments were repeated at least 3 times. After 48 h transfection, Western blot analysis was performed as above, the primary antibodies were mouse anti-p27 (NeoMarkers, Fremont, CA, USA, 1:500 dilution), mouse anti-c-Myc (Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:500 dilution), and mouse anti-Bcl-2 (NeoMarkers, Fremont, CA, USA, 1:500 dilution), rabbit anti-proliferating cell nuclear antigen (PCNA) (NeoMarkers, Fremont, CA, USA, 1:1000 dilution), mouse anti-survivin (Chemicon, Temecula, CA, USA, 1:1000 dilution) and mouse anti-β-actin (Sigma, 1:20000 dilution).
Statistical analysis All data were expressed as mean±SD. Statistical analysis was performed by the Student’s t-test. P<0.05 was indicated to be statistical significant.
Results
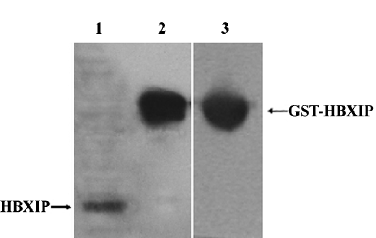
Identification of antibody of rabbit anti-HBXIP To examine HBXIP, we generated rabbit anti-HBXIP antibody recognizing HBXIP. Western blot analysis showed that this anti-HBXIP antibody specifically recognized HBXIP in MCF-7 cells, and the GST-HBXIP fusion protein expressed in Escherichia coli BL21 (DE3) and GST in the fusion protein GST-HBXIP from BL21 (DE3) cells could also be detectable using antibody of anti-GST (Figure 1).
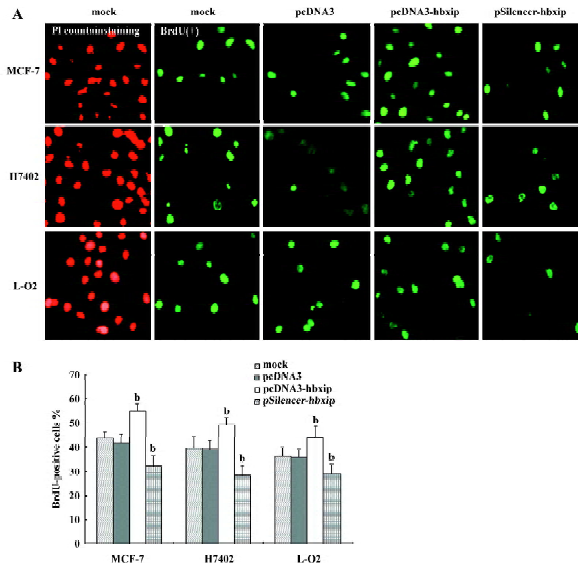
Promotion of cell proliferation by HBXIP overexpression Transfection efficiency revealed that approximately 70%–80% of cells showed green fluorescence (Figure 2). To evaluate whether HBXIP expression correlated with fundamental cellular processes, we investigated the cell proliferation by MTT assay, BrdU incorporation assay, flow cytometry analysis, and Western blot analysis after transfection. MTT assay showed that transfection with pcDNA3-hbxip plasmid promoted cell proliferation (P<0.05 vs control), whereas transfection with the pSilencer-hbxip plasmid decreased cell proliferation by MTT assay in MCF-7 cells (P<0.05 vs control, data not shown). To further confirm the results, we repeated the same experiments in hepatoma H7402 cells and L-O2 cells. BrdU incorporation analysis showed that the induction of DNA synthesis by HBXIP was assessed in MCF-7, H7402, and L-O2 cells. The results showed that the percentage of cells in the S phase increased significantly in MCF-7, H7402, and L-O2 cells transfected with pcDNA3-hbxip compared with the cells transfected with pcDNA3 (P<0.05), and was reduced significantly in cells transfected with pSilencer-hbxip plasmids (Figure 3). No statistically significant difference was observed between the control cells and cells transfected with pcDNA3 empty vector.
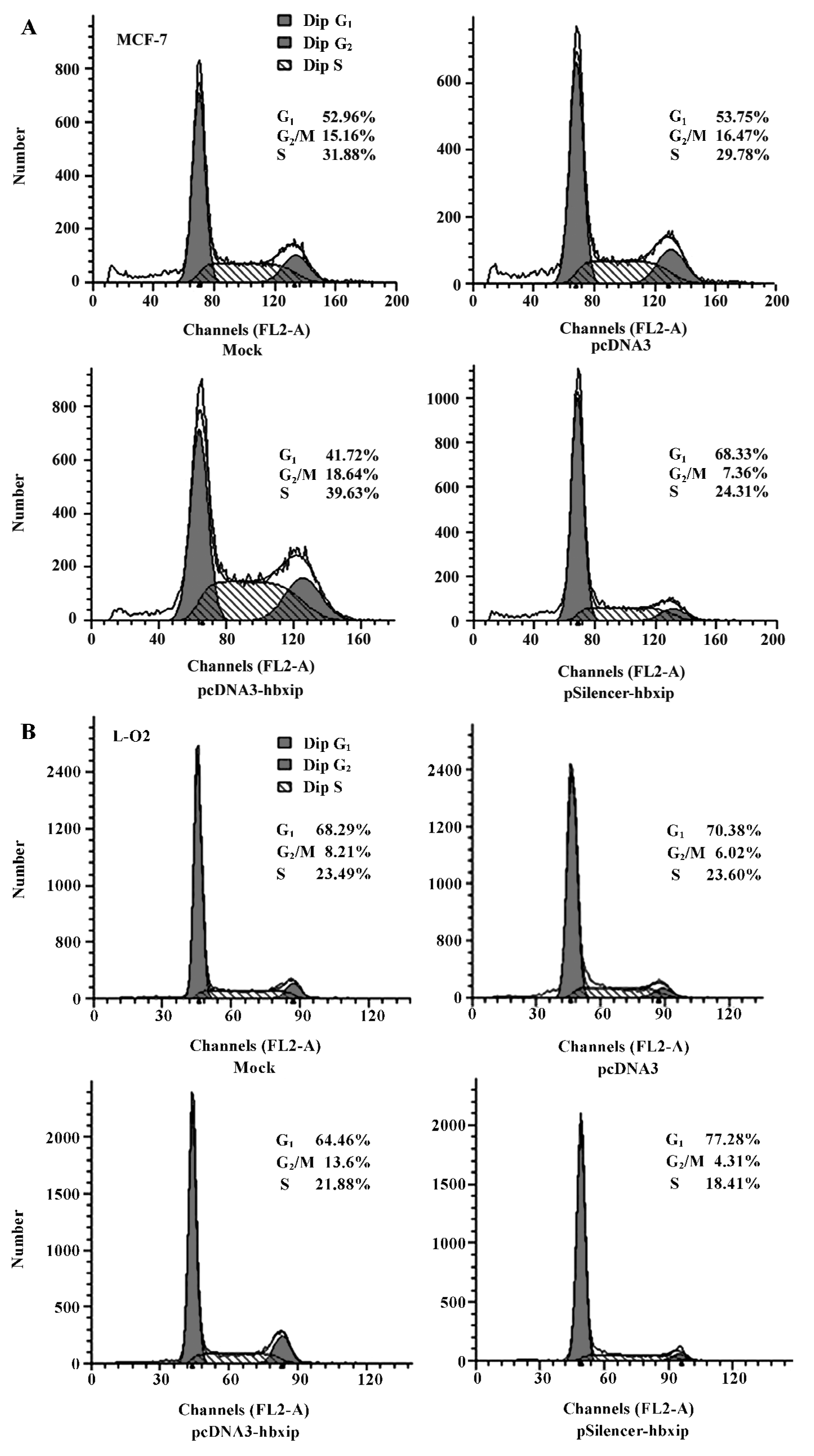
HBXIP overexpression significantly accelerated the cell proliferation compared with untransfected and pcDNA3 transfected cells. Cell proliferation was quantified by PI. PI is the sum of the S and G2/M phase activities of the cell cycle expressed as a fraction of the total cell population, that is, PI=[(S+G2/M)/(G0/G1+S+G2/M)]×100%. Flow cytometry analysis showed that overexpression of HBXIP, transfected with pcDNA3-hbxip plasmids, led to an increased cell PI from 46.25% to 58.28% in MCF-7 cells. However, the downregula-tion of HBXIP, transfected with pSilencer-hbxip plasmid, resulted in a decreased cell PI (from 46.25% to 31.67%) and increased percentage of cells in the G1 phase (from 53.75% to 68.33%, P<0.01; Figure 4A). Flow cytometry analysis was also repeated in L-O2 cells (Figure 4B), and the PI of the L-O2 cells transfected with pcDNA3-hbxip plasmids increased significantly compared with the pcDNA3 control (from 29.62% to 35.54%, P<0.01). The PI in pSilencer-hbxip transient transfection cells was only 22.72%, so HBXIP also accelerated the cell proliferation of L-O2 cells. Flow cytometry was repeated 3 times.
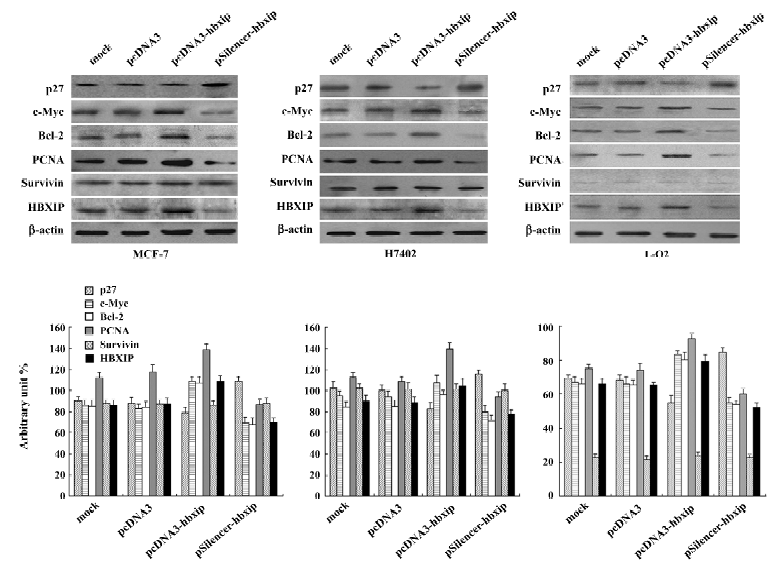
Involvement of proteins in the promotion of cell proliferation To investigate the mechanism, we examined some proteins related to cell proliferation and cell-cycle regulation, such as c-Myc, Bcl-2, PCNA, and p27. Western blot analysis showed that transient transfection with pcDNA3-hbxip plasmid upregulated expression of c-Myc, Bcl-2, and PCNA, but downregulated p27 in MCF-7, H7402, and L-O2 cells. However, RNAi targeting HBXIP mRNA with the pSilencer-hbxip plasmid reduced the expression levels of c-Myc, Bcl-2, PCNA, whereas the p27 expression level was upregulated with the downexpression of HBXIP in these cells. Since HBXIP was a cofactor for survivin, we also detected the expression of survivin in the same condition. The expression level of survivin was not affected by the overexpression or depression of HBXIP, which was consistent with the previous report[2]. We further confirmed this finding by applying Glyco Band-Scan software (PROZYME, San Leandro, CA, USA; Figure 5).
Discussion
The functions of HBXIP were investigated in breast cancer MCF-7 cells, hepatoma H7402 cells, and the normal hepatic cell line, L-O2. We overexpressed HBXIP in the above cells by transfection with the pcDNA3-hbxip plasmid and followed with an investigation of cell proliferation by MTT, BrdU incorporation assay, and flow cytometry analysis. The findings showed that the overexpression of HBXIP was able to promote cell proliferation. The depression of cell proliferation was found by RNAi targeting HBXIP mRNA in the cells. As we know, cell proliferation is functionally linked to the expression of genes associated with growth control. The maintenance of normal cell function and tissue homeostasis is depend on the precise regulation of multiple signaling pathways that control cellular decisions to either proliferate, differentiate, arrest cell growth, or apoptosis. Previous studies indicated that HBXIP was a necessary cofactor of survivin in the process of suppression of apoptosis in cancer cells, and increased HBXIP was found in both cancerous and non-malignant liver tissue of humans with chronic HBV infection[2], so we supposed that the function of HBXIP might relate to the cell proliferation.
Marusawa et al reported that HBXIP was a link in bridging HBX and survivin[2]. HBX plays an important role in the development of liver disease and exhibits effects on gene transcription, cell proliferation, survival, and apoptosis[9,10]. It stimulates cell-cycle progression, shortening the emergence of cells from quiescence (G0) and entry into the S phase by stimulating the Ras-signaling pathway, and accelerating transition through checkpoint controls at the G0/G1 and G2/M phases[11]. Survivin is a member of IAP (inhibitor of apoptosis protein) family and has been implicated in anti-apoptosis, cell division, and cell-cycle control[12–15]. Under normal physiological conditions, survivin is involved in coordinating chromosomes and mitosis[16].
In the present study, our data indicated that overexpression of HBXIP resulted in more MCF-7 cells going into the S phase of the cell cycle. p27 is a member of the Cip/Kip family of cyclin-dependent kinase inhibitor (CDKI) that regulate cell-cycle progression, thus inhibiting various cycle-CDK complexes. Physiologically, p27 is believed to primarily regulate the progression of cells from late G1 into the S phase by interacting with the cyclin E-CDK2 complex[17,18]. We found that transient transfection with the pcDNA3-hbxip plasmid upregulated the expression of c-Myc, Bcl-2, and PCNA, but downregulated the expression of p27 in these cell lines. c-Myc is a nuclear phosphoprotein that functions as a transcription factor stimulating both cell-cycle progression and apoptosis. It plays a critical role in normal cell-cycle progres-sion, especially during transition from G1 to the S phase[19]. c-Myc is also an early response gene, which responds directly to mitogenic signals to push cells in the G1 phase of the cell cycle[20]. The proto-oncoprotein Bcl-2 is a powerful antagonist of the mitochondrial pathway of apoptosis initiated by a variety of extra- and intra-cellular stresses. As the Bcl-2 family members reside upstream of irreversible cellular damage and focus much of their efforts on the level of mitochondria, they play a pivotal role in deciding whether a cell will live or die[21]. PCNA is a highly conserved 36 kDa acidic nuclear protein that is expressed during cell replication and DNA repair, and is correlated with cell proliferation[22]. The same results, that is, upregulation of c-Myc, Bcl-2, and PCNA, were also observed in H7402 cells and L-O2 cells by overexpression of HBXIP.
Protein-protein interactions are crucial for all biological processes. Using systematic, automated, large-scale Y2H matrix interaction mating, Ulrich Stelzl et al[23] screened other interacting proteins of HBXIP, such as eukaryotic translation elongation factor 1 alpha 1 (EEF1A1), glyceraldehyde-3-phosphate dehydrogenase (GAPD), G protein-coupled receptor kinase interactor 1 (GIT1), KIAA1377, and rap1-interacting factor 1 (RIF1). These proteins in cellular functions are different. Therefore, HBXIP may also contribute more besides its effects on apoptosis and cell proliferation. Additionally, the expression of HBXIP could be regulated by some factors, such as garlic[24], gonadotropins, and forskolin[25].
Taken together, our findings indicate that one of the functions of HBXIP is its involvement in proliferation regulation in cancer cell lines and the normal liver cell line, which is related to cell-cycle transition through checkpoint controls at the G0/G1 or G2/M phases and the downregulation of p27. In addition, the upregulation of c-Myc by HBXIP may also play an important role in cell-cycle transition.
References
- Melegari M, Scaglioni PP, Wands JR. Cloning and characterization of a novel hepatitis B virus X binding protein that inhibits viral replication. J Virol 1998;72:1737-43.
- Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I, et al. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J 2003;22:2729-40.
- Fujii R, Zhu C, Wen Y, Marusawa H, Bailly-Maitre B, Matsuzawa S, et al. HBXIP, cellular target of hepatitis B virus oncoprotein, is a regulator of centrosome dynamics and cytokinesis. Cancer Res 2006;66:9099-107.
- Minczuk M, Mroczek S, Pawlak SD, Stepien PP. Human ATP-dependent RNA/DNA helicase hSuv3p interacts with the cofactor of survivin HBXIP. FEBS J 2005;272:5008-19.
- Zhang X, Ma H, Ye L, Dong N, Shi Z, Sha L, Wang H. Effect of hepatitis B X interacting protein on apoptosis induced by hepatitis B virus X protein. Chin J Biochem & Mol Bio 2005;21:403-7.
- Zhang X, Dong N, Yin L, Cai N, Ma H, You J, et al. Hepatitis B virus X protein upregulates survivin expression in hepatoma tissues. J Med Virol 2005;77:374-81.
- Park YO, Hwang ES, Moon TW. The effect of lycopene on cell growth and oxidative DNA damage of Hep3B human hepatoma cells. Biofactors 2005;23:129-39.
- Chen Z, Li DQ, Tong L, Stewart P, Chu C. P flugfelder SC. Targeted inhibition of p57 and p15 blocks transforming growth factor beta-inhibited proliferation of primary cultured human limbal epithelial cells. Mol Vis 2006;12:C983-94.
- Madden CR, Slagle BL. Stimulation of cellular proliferation by hepatitis B virus X protein. Dis Markers 2001;17:153-7.
- Murakami S. Hepatitis B virus X protein: a multifunctional viral regulator. J Gastroenterol 2001;36:651-60.
- Benn J, Schneider RJ. Hepatitis B virus HBx protein deregulates cell cycle checkpoint controls. Proc Natl Acad Sci USA 1995; 92: 11 215–9.
- Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997;3:917-21.
- Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998;396:580-4.
- Suzuki A, Shiraki K. Tumor cell “dead or alive”: caspase and survivin regulate cell death, cell cycle and cell survival. Histol Histopathol 2001;16:583-93.
- Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003;22:8581-9.
- Vagnarelli P, Earnshaw WC. Chromosomal passengers: the four-dimensional regulation of mitotic events. Chromosoma 2004;113:211-22.
- Chiarle R, Pagano M, Inghirami G. The cyclin dependent kinase inhibitor p27 and its prognostic role in breast cancer. Breast Cancer Res 2001;3:91-4.
- Lloyd RV, Erickson LA, Jin L, Kulig E, Qian X, Cheville JC, et al. p27KIP1: a multifunctional cyclin-dependent kinase inhibitor with prognostic significance in human cancers. Am J Pathol 1999;154:313-23.
- Spencer CA, Groudine M. Control of c-myc regulation in normal and neoplastic cells. Adv Cancer Res 1991;56:1-48.
- de Alboran IM, O’Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, et al. Analysis of c-myc function in normal cells via conditional gene-targeted mutation. Immunity 2001;14:45-55.
- Gross A, McDonnell JM, Korsmeyer SJ. Bcl-2 family members and the mitochondria in apoptosis. Genes & Development 1999;13:1899-911.
- Shivji MKK, Kenny MK, Wood RD. Proliferating cell nuclear antigen is required for DNA excision repair. Cell 1992;69:367-74.
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005;122:957-68.
- Li Y, Lu YY. Applying a highly specific and reproducible cDNA RDA method to clone garlic up-regulated genes in human gastric cancer cells. World J Gastroenterol 2002;8:213-6.
- Rimon E, Sasson R, Dantes A, Land-Bracha A, Amsterdam A. Gonadotropin-induced gene regulation in human granulosa cells obtained from IVF patients: modulation of genes coding for growth factors and their receptors and genes involved in cancer and other diseases. Int J Oncol 2004;24:1325-38.