
Pharmacokinetics and tissue distribution of a novel PDE5 inhibitor, SK-3530, in rats1
Introduction
Male erectile dysfunction (MED), the persistent inability to achieve or maintain an erection for satisfactory sexual performance, is a common and important medical problem. Sildenafil is a potent and selective inhibitor of the human cGMP-specific phosphodiesterase type 5 (PDE5), the predominant isozyme responsible for the metabolism of cGMP in the corpus cavernosum of the penis[1–4]. The development of sildenafil citrate (Viagra) as an effective and orally active agent for the treatment of MED revolutionized the treatment of this disease[5,6]. Despite the efficacy of sildenafil as a treatment for MED, there are some notable drawbacks associated with its use. Clinically significant adverse effects, such as headache, facial flushing, dyspepsia, and visual disturbance have been reported[1,7,8].
5-Ethyl-2-{5-[4-(2-hydroxy-ethyl)-piperazine-1-sulfonyl]-2-propoxy-phenyl}-7-propyl-3,5-dihydro-pyrrolo(3,2-d)pyrimidin-4-one (SK-3530; Figure 1) is a novel PDE5 inhibitor that was synthesized to alleviate the drawbacks of sildenafil, and is now being developed as a new drug candidate. Herein, we describe the pharmacokinetics and tissue distribution of SK-3530 in rats using the radioisotope-labeled compound as part of the pharmacological preclinical study of SK-3530.
Materials and methods
Chemicals and reagents 14C-labeled SK-3530 (specific activity 93.52 µCi/mg) was synthesized at Korea Radiochemicals Center (Suwon, Korea) with radiochemical purity ≥98% as judged by HPLC-radiochromatography. Unlabeled SK-3530 was provided by SK Chemical Ltd (Gunpo, Korea) with a chemical purity of 99%. Polyethylene glycol (PEG, average molecular weight 300), sodium hydroxide (NaOH), and ammonium formate were obtained from Sigma Chemical Co (St Louis, MO, USA). Ethyl acetate (EtOAc), acetonitrile, and methanol were of HPLC grade and purchased from Mallinckrodt Baker (Phillipsburg, NJ, USA).
Animals Male Sprague-Dawley rats weighing 210–250 g were purchased from DaeHan Laboratory Animal Research Center (Taejeon, Korea). The animals were housed at 23±2 ºC temperature and moisture [55%±10%]-controlled room, with a 12 h light/dark cycle, and allowed free access to food and water.
Preparation of dosing solution Appropriate quantities of 14C-labeled SK-3530 were diluted with cold SK-3530 to adjust the specific activity required for the dose preparation (6 µCi for pharmacokinetics and 2 µCi for tissue distribution). SK-3530 was dissolved in PEG solution (50% in water).
Pharmacokinetic experiment Two days before adminis-tration, the femoral artery and vein intravenous (iv) only were cannulated using polyethylene-50 and polyethylene-10 tubing (Becton Dickinson, Lincoln Park, NJ, USA). The cannulae were fixed to the back of the neck. The rats were fasted overnight before drug administration and until 6 h after dosing. For the iv experiment, the rats (n=4) were given a single 5 mg/kg bolus of 14C-labeled SK-3530. Heparinized samples of blood (0.4 mL) were collected at 1, 5, 10, 30, 60, 90, 120, 180, 240, 360, 480, and 600 min post-dose. For the oral experiments, the rats (n=4) were given a single dose of 10, 20, and 40 mg/kg, respectively. Then the blood samples were collected at 10, 20, 30, 60, 90, 120, 180, 240, 360, 480, 600, and 720 min post-dose. Plasma was harvested after centrifugation and stored frozen at -20 ºC until analyzed.
Distribution The rats (n=4 per group) were dosed with [14C]SK-3530 orally at 40 mg/kg. At 1, 4, and 24 h after dosing, each animal was lightly anaesthetized with diethyl ether. Blood was collected by heart puncture and the rats were then killed by cervical dislocation. The representative tissues or organs were rapidly dissected, washed with saline, weighed, and patted dry on a combustion pad in preparation for sample oxidation.
Radioactivity measurements Plasma was mixed with 10 mL scintillation fluid (Insta Gel XF, Packard, Meriden, CT, USA) and counted directly for radioactivity. Aliquots of tissue samples were weighed and combusted in a sample oxidizer (Tri-Carb Model 307, Packard, Meriden, CT, USA). The resulting 14CO2 was absorbed on Carbosorb (Packard, Meriden, CT, USA) and then mixed with Permafluor V scintillation fluid (Packard, Meriden, CT, USA) The radioactivity of samples was counted using a liquid scintillation counter (Packard, USA).
LC-MS/MS analysis To the 100 µL plasma sample we added 5 µL internal standard solution (sildenafil 2 mg/L in methanol), 100 µL of 50 mmol/L NaOH, and 700 µL EtOAc. After voltex-mixing and centrifugation, the supernatant was dried under nitrogen evaporation. The residue was dissolved in HPLC initial buffer and then analyzed by LC-MS/MS. The LC-MS/MS system consisted of a LC-10ADvp binary pump system with an API2000 triple quadruple mass spectrometer (Applied Biosystems-SCIEX, Concord, Canada) equipped with a turboionspray source. The HPLC mobile phases consisted of 5 mmol/L ammonium formate buffer (pH 6.0) and 5 mmol/L ammonium formate in 90% acetonitrile. Chromatographic separation was achieved on a Capcellpak Phenyl (1.5×150 mm, 5 µm, Shiseido, Tokyo, Japan) using a isocratic elution of 80% solvent at a flow rate of 0.15 mL/min. The run time was 3.5 min. Electrospray ionization was performed in the positive mode with nitrogen as the nebulizing turbo spray and curtain gas, with optimum values set at 40, 80, and 40 (arbitrary units). Multiple reaction monitoring (MRM) detection was employed using nitrogen as the collision gas (4 arbitrary units) with a dwell time of 150 ms for each transition; the transitions monitored were 532→99 for SK-3530 and 475→58 for sildenafil (IS). The representative MRM chromatograms for SK-3530 and sildenafil in the rat plasma sample are presented in Figure 2.
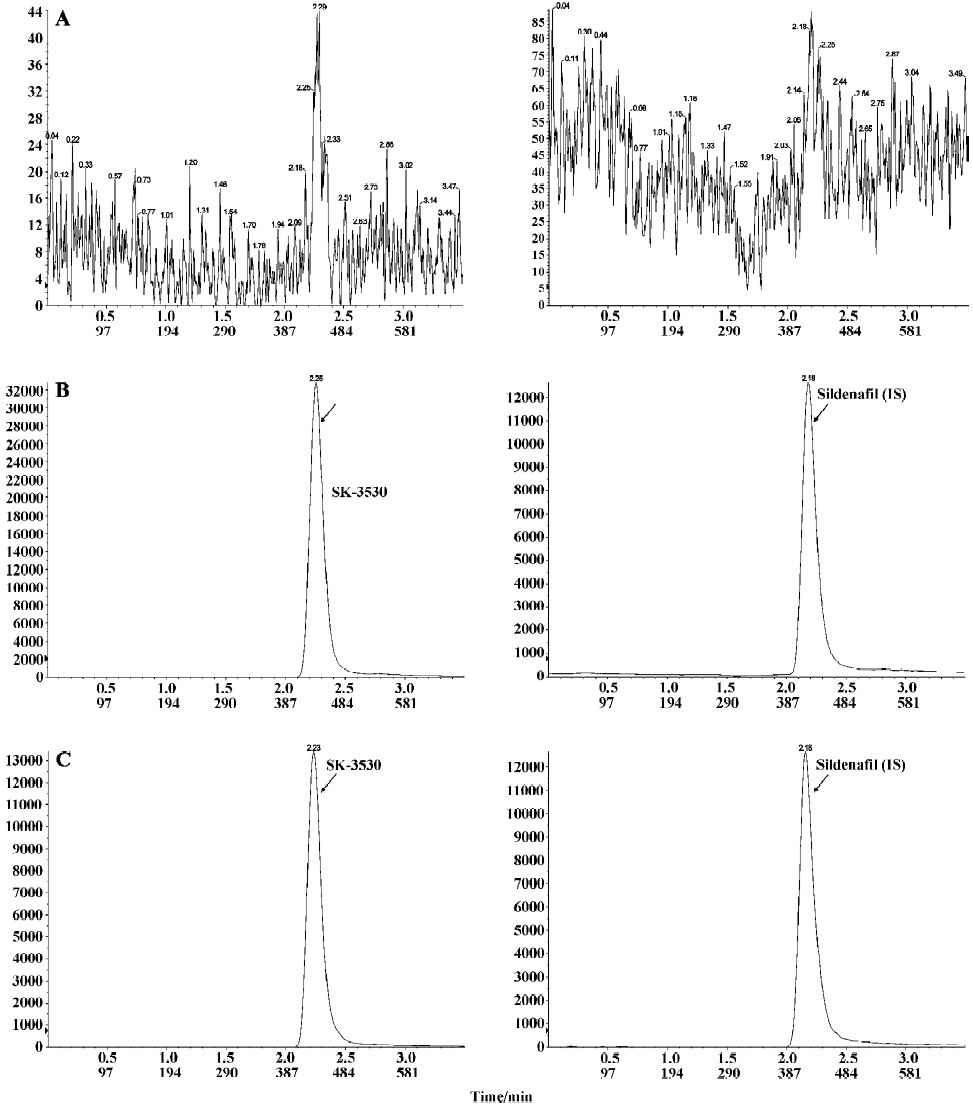
The calibration standards for SK-3530 were prepared by adding various amounts of SK-3530 to 100 µL rat blank serum and analyzed in the same manner as the plasma samples from the dosed rats. The calibration curves were prepared by plotting peak area ratios of SK-3530/sildenafil (IS) against the SK-3530 concentration and analyzed by the linear least-squares regression analysis. The calibration curve for SK-3530 was linear over the concentration range of 5–5000 µg/L with correlation coefficients (r2) more than 0.99.
For the validation of the LCMS/MS method, precision and accuracy were determined by repeated analysis of 3 concentration levels of quality control samples (5, 200, and 5000 µg/L, n=3) on 3 separate days. The limit of quantification was 5 µg/L. The accuracy of the method was more than 90%. The inter-day precisions (as the percentage relative standard deviation) were 10.2%, 0.7%, and 4.2% at 5, 200, and 5000 µg/L, respectively).
Pharmacokinetic analysis The area under the plasma concentrationtime curve (AUC), total plasma clearance, terminal half-life, and volume of distribution at steady-state (Vdss) were determined by a non-compartmental method using WinNonlin (Scientific Consulting, Lexington, KY, USA). Maximum plasma concentration (Cmax) and the corresponding Tmax were reported as observed. Oral bioavailability was calculated as follow:
Bioavailability (%)=(AUCoral/doseoral)×(doseiv/AUCiv)×100.
Protein binding assay Plasma protein binding was determined by ultrafiltration using Amicon centrifuge micropartition devices (Amicon, Beverly, MA, USA; molecular weight cut-off, 30 000). [14C]SK-3530 was added to the blank rat and human plasma samples (n=4) to yield final concentrations of 1, 5, 50, and 100 µmol/L and incubated for 10 min at 37 °C. After incubation, the mixtures were added to the ultrafiltration units and centrifuged at 37 °C for 30 min at 14 000×g. The concentrations of [14C]SK-3530 in the plasma and ultrafiltrate were determined by liquid scintillation spectrometry. For the ex vivo determination of plasma protein binding, a 40 mg/kg dose of [14C]SK-3530 was orally administered and blood samples were drawn at 1, 4, and 24 h after dosing. The plasma samples were then prepared and protein binding was analyzed as described earlier.
Results
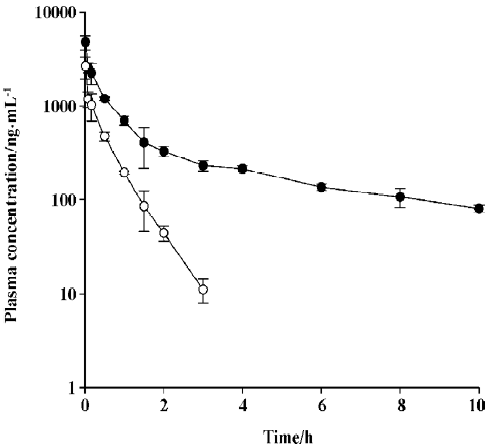
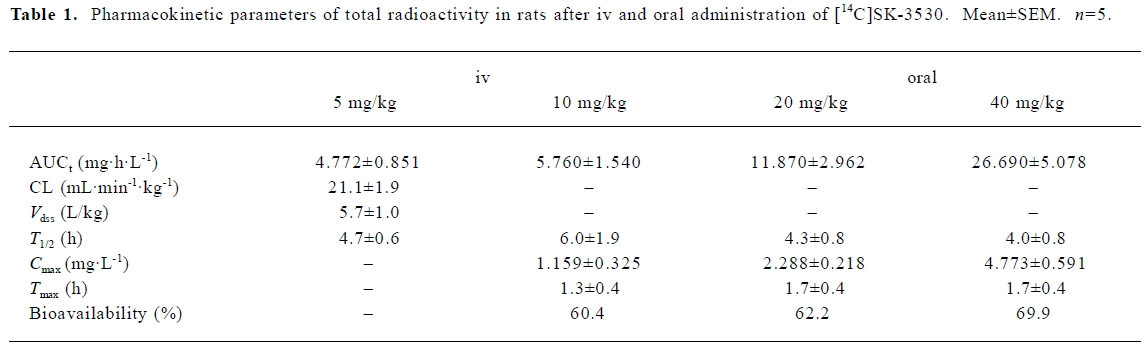
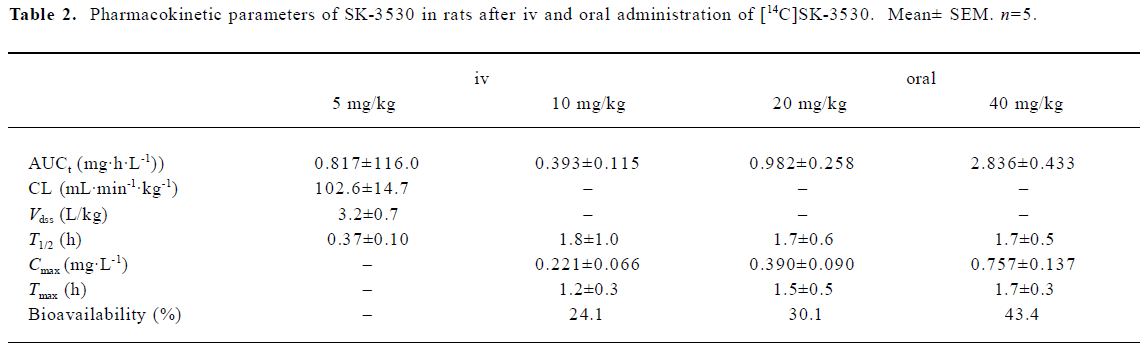
Plasma concentrations of total radioactivity and SK-3530 The mean arterial plasma concentration-time profiles of total radioactivity and SK-3530 after iv administration at a dose of 5 mg/kg in the rats are shown in Figure 3. After iv administration of [14C]SK-3530, the parent SK-3530 was rapidly cleared with a terminal half-life of 0.37 h and was no longer detected at 4 h after dosing, whereas the radioactive equivalents in the plasma declined more slowly. Total radioactivity was eliminated in a biphasic fashion with a terminal half-life of 4.7 h. The AUC of the parent SK-3530 was only 17% compared with the AUC of the total radioactivity. The Vdss was higher than the volume of body water at 5.7 and 3.2 L/kg for the total radioactivity and parent SK-3530, respectively. Clearance was measured as 21.1 and 102 mL·min-1·kg-1 for the total radioactivity and parent SK-3530, respectively.
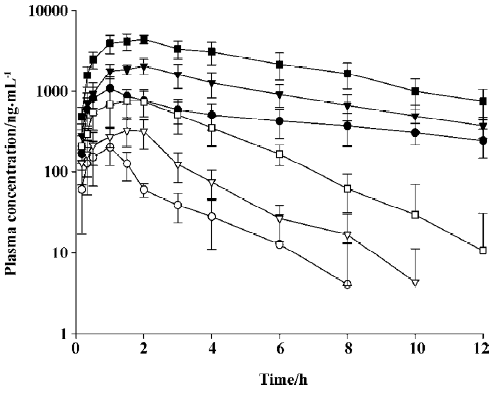
The mean arterial plasma concentration-time profiles of the total radioactivity and SK-3530 following oral administration at doses of 10, 20, and 40 mg/kg in the rats are shown in Figure 4. After the oral administration of [14C]SK-3530, the drug was absorbed; the peak concentration of the total radioactivity occurred within 1.3–1.7 h. The parent SK-3530 peaked at 1.2–1.7 h, which was similar to the total radioactivity profile, but declined more rapidly. Increases in the Cmax and AUC of the total radioactivity were almost proportional to the administered dose, suggesting that SK-3530 has linear pharmacokinetics. However, in the case of the arent SK-3530, the AUC increased at disproportionately higher rates compared to the administered doses. These phenomena seem to be due to metabolic saturation at the higher SK-3530 doses. The AUC of the parent compound was 6.8%–10.6% compared with that of the total radioactivity profile. The oral bioavailabilities based on the total radioactivity were estimated to be 60.4%, 62.2%, and 69.9% for 10, 20, and 40 mg/kg doses, respectively. The oral bioavailabi-lities of the parent SK-3530 were estimated to be 24.1%, 30.1%, and 43.4% for 10, 20, and 40 mg/kg doses, respectively. The pharmacokinetic parameters of the total radioactivity and parent SK-3530 are summarized in Tables 1 and 2.
Full table
Full table
Plasma binding The in vitro incubation of [14C]SK-3530 with rat and human plasma samples resulted in high protein binding rates of more than 97%; binding was concentration-independent over the range tested (1–100 µmol/L), with no difference between the species show. An in vivo plasma analysis revealed that more than 98% of the radioactivity (98.3%, 99.0%, and 98.5% for samples collected at 1, 4, and 24 h post-dose, respectively) was associated with the plasma protein.
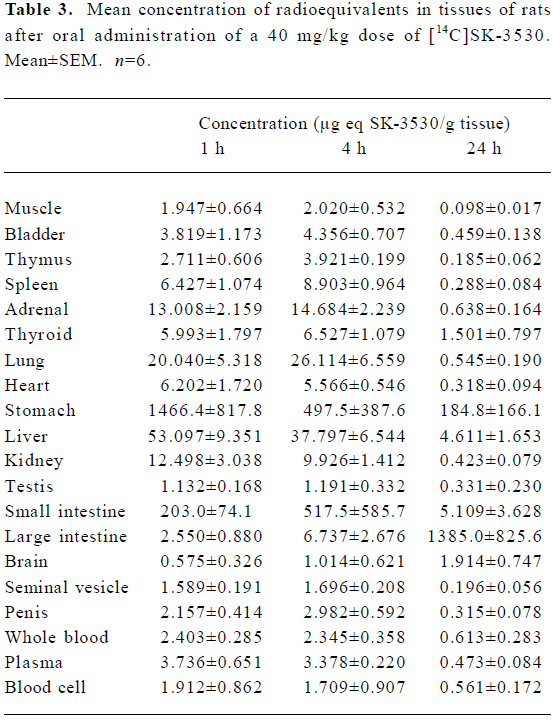
Tissue distribution The tissue distribution of the total radioactivity after a single oral administration of [14C]SK-3530 at a dose of 40 mg/kg is shown in Table 3. Radioactivity was widely distributed into all tissues. The tissue/plasma radioactivity ratio at 1 h after administration was calculated to be 0.15–5.4 with the exception of the excretory organs. The concentrations of the total radioactivity for several representative tissues are as follows: 13.0 µg equivalents/g in the adrenal glands, 20.0 µg equivalents/g in the lungs, 53.1 µg equivalents/g in the liver, 6.2 µg equivalents/g in the heart, and 12.5 µg equivalents/g in the kidneys. At 4 h post-dose, the radioactivity of several tissues slightly increased or decreased, but on the whole, no significant changes of radioactivity were shown compared to the radioactivity profile at 1 h post-dose. The radioactivity decreased in most tissues over a 24 h period, but was still detectable with relatively high penetration shown in the thyroid, liver, and brain. Notably, the radioactivity of the brain gradually increased with time up to 24 h post-dose.
Full table
Discussion
After the oral administration of [14C]SK-3530 in the rats, the maximum plasma concentration of the total radioactivity was achieved around 1 h post-dose, showing a moderate absorption rate of SK-3530 from the gastrointestinal tract. The Cmax and AUC of the total radioactivity increased almost proportionally to the dose, indicating that SK-3530 has linear kinetics over the dose range of 10–40 mg/kg. However, the AUC of the parent SK-3530 showed a hyperbolic pattern with increasing doses, suggesting that metabolic saturation occurred with high orally administered doses.
The oral bioavailability, based on the total radioactivity, was estimated to be between 60% and 70%, whereas that of the parent SK-3530 was calculated to be between 20% and 40%, suggesting a considerable amount of SK-3530 was eliminated through an extensive first-pass metabolism, such as sildenafil[9,10]. The low bioavailability of sildenafil after oral administration has been reported to be mainly due to a considerable intestinal first-pass effect[11], which can consequently be considered as a major cause for the low bioavaila-bility of SK-3530 considering their structural similarity. SK-3530 is modified from sildenafil at N- or O-side chain and has a dihydropyrrole ring instead of a pyrazole ring. It was reported that the chemical modifications in SK-3530 enhanced specific activity for PDE5 inhibition and consequently reduced adverse effects[12]. The principle metabolic pathways of sildenafil is known to be piperazine N-demethylation, pyrazole N-demethylation, piperazine N,N'-deethylation, oxidation of the piperazine ring, and aliphatic hydroxyla-tion[10]. In the metabolism study of SK-3530 using LC-MS/MS (unpublished data), SK-3530 exhibited almost a similar metabolic pattern to that of sildenafil, except for pyrazole N-dealkylation and conjugates formation. Significant differences, especially, the enhancement of bioavailability, were not observed in SK-3530 pharmacokinetic data compared with that of sildenafil. It seems that the pharmacokinetic properties of SK-3530 make little contribution to the enhanced compliance of SK-3530.
SK-3530 exhibits a volume of distribution (Tables 1 and 2) in excess of the volume of body water (~0.8 L/kg), indicating the high tissue affinity of SK-3530, which is in keeping with the weak, basic nature of SK-3530. The tissue distribution data showed the [14C]SK-3530 radio equivalents were well distributed in all the tissues examined. The tissue/plasma radioactivity ratio at 1 h after administration was calculated to be 0.15–5.4, with the exception of the excretory organs, indicating that [14C]SK-3530 radio equivalents have relatively high tissue affinity. The highest concentration of radio equivalents was found in the gastrointestinal tract, resulting from either the unabsorbed drug following oral administration or biliary elimination of drug radio equivalents. Notably, the radioactivity of brain tissue gradually increased with the incubation time up to 24 h post-dose, suggesting [14C]SK-3530 radio equivalents cross the blood–brain barrier and potentially exert an inhibitory effect on PDE5 in the cerebral blood vessels.
[14C]SK-3530 exhibited a high protein binding rate. Nevertheless, the actual effect of protein binding on either the total drug distribution to tissues or the drug levels in tissues is considered minimal, as at least 97% of the dose resides were outside the circulation, that is, within the tissues, based on the volume of SK-3530 distribution (~3.2 L/kg) and the blood volume of the rats (~0.1 L/kg)[13].
In conclusion, the present study has demonstrated that the administered SK-3530 was relatively well absorbed in the gastrointestinal tract and showed linear pharmacokinetics over the investigated dose range. SK-3530 had low oral bioavailability due to a high first-pass metabolism. The moderate lipophilic and weak, basic natures of SK-3530 result in extensive tissue distribution and clearance due to metabolism.
References
- Boolell M, Allen MJ, Ballard SA, Gepi-Attee S, Muirhead GJ, Naylor AM, et al. An orally active type 5 cyclic GMP specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int J Impotence Res 1996;8:47-52.
- Carter AJ, Ballard SA, Naylor AM. Effect of the selective phosphodiesterase type 5 inhibitor sildenafil on erectile dysfunction in the anesthetized dog. J Urol 1998;160:242-6.
- Corbin JD, Francis SH, Webb DJ. Phosphodiesterase type 5 as a pharmacologic target in erectile dysfunction. Urology 2002;60:4-11.
- Rosen RC, Kostis JB. Overview of phosphodiesterase 5 inhibition in erectile dysfunction. Am J Cardiol 2003;92:9M-18M.
- Boolell M, Gepi-Attee S, Gingell JC, Allen MJ. Sildenafil, a novel effective oral therapy for male erectile dysfunction. Br J Urol 1996;78:257-61.
- Kloner RA. Cardiovascular effects of the 3 phosphodiesterase-5 inhibitors approved for the treatment of erectile dysfunction. Circulation 2004;110:3149-55.
- Kruuse C, Thomsen LL, Jacobsen TB, Olesen J. The phosphodiesterase 5 inhibitor sildenafil has no effect on cerebral blood flow or blood velocity, but nevertheless induces headache in healthy subjects. J Cereb Blood Flow Metab 2002;22:1124-31.
- Kruuse C, Frandsen E, Schifter S, Thomsen LL, Birk S, Olesen J. Plasma levels of cAMP, cGMP and CGRP in sildenafil-induced headache. Cephalalgia 2004;24:547-53.
- Nichols DJ, Muirhead GJ, Harness JA. Pharmacokinetics of sildenafil after single oral doses in healthy male subjects: absolute bioavailability, food effects and dose proportionality. Br J Clin Pharmacol 2002;53:5S-12S.
- Walker DK, Ackland MJ, James GC, Muirhead GJ, Rance DJ, Wastall P, et al. Pharmacokinetics and metabolism of sildenafil in mouse, rat, rabbit, dog and man. Xenobiotica 1999;29:297-310.
- Shin HS, Bae SK, Lee MG. Pharmacokinetics of sildenafil after intravenous and oral administration in rats: Hepatic and intestinal first-pass effects. Int J Pharm 2006;320:64-70.
- Shin HI, Lee J, Kim DK. Synthesis of 5-ethyl-2-{5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]-2-n-propoxyphenyl}-7-n-propyl-3,5-dihydro-4H-pyrrolo[3,2-d]-[2-14C]pyrimidin-4-one ·2HCl (14C-SK3530·2HCl). J Label Compd Radiopharm 2006;49:1141-9.
- Diehl KH, Hull R, Morton D, Pfister R, Rabemampianina Y, Smith D, et al. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol 2001;21:15-23.