
Blockade of sonic hedgehog signal pathway enhances antiproliferative effect of EGFR inhibitor in pancreatic cancer cells1
Introduction
Pancreatic carcinoma is characterized by its aggressive local invasion of adjacent structures. At the time of first diagnosis, only 10%–20% of cases are eligible for the potentially curative Whipple’s procedure. Furthermore, pancreatic cancer is relatively resistant to both chemotherapy and radiotherapy[1]. Further understanding of the biological roles of the genotypic changes during pancreatic carcinogenesis may provide new clues for developing strategies to prevent and treat this disease.
Among the various genotypic changes occurring in pancreatic cancer, the dysregulation of the sonic hedgehog (SHH) signaling pathway has been reported[2]. The SHH protein is expressed in an embryo and participates in regulating cell proliferation, differentiation, and tissue patterning of many organs, including pancreas. The SHH ligand mediates its biological effects through the multi-component receptor complex constituted from a transmembrane protein PATCHED-1 (PTCH-1). PTCH-1 binds SHH with high affinity and Smoothened (SMO), a second signaling transmembrane G protein-coupled receptor. In the absence of SHH, PTCH-1 represses SMO activity, while the binding of SHH to PTCH-1 releases the basal repression of SMO by PTCH-1. This ultimately leads to the activation of the glioma-associated oncogene homolog 1(GLI-1) transcription factor, which induces the expression of numerous target genes that regulate proliferation, differentiation, and extracellular matrix interactions[3]. Recently, this signaling pathway has been shown to have some interactions with other pathways, such as the PI3-kinase/Akt signaling pathways[4], and plays a major role in several types of gastrointestinal cancers[5]. The abnormal activation of the SHH pathway in carcinogenesis makes it a potential target for therapy. Another pathway that may play an important role in pancreatic carcinogenesis is the family members of epidermal growth factor receptor(EGFR) and their ligand molecules. The overexpression of EGFR signaling increases the proliferation of pancreatic cancer cells[6].
While the expression of SHH and EGFR has been reported in many tumors, respectively, the relationship between these signaling pathways was unclear. Moreover, few studies have been reported about the co-expression and simultaneous inhibition of SHH and EGFR signaling in pancreatic cancer cells. The aim of the present study was to detect the elements of the SHH and EGFR pathways in 3 pancreatic cancer cell lines, and to determine whether the simultaneous inhibition of SHH and EGFR pathways might be more effective than single agents. Hence, the antiprolifera-tive effects induced by cyclopamine, a SHH signaling specific inhibitor, alone or in combination with EGFR inhibitor Iressa, were investigated in the PANC-1, SUIT-2, and ASPC-1 pancreatic cancer cell lines, and the interaction between these 2 pathways was evaluated.
Materials and methods
Cell culture Pancreatic cancer cell lines (PANC-1, SUIT-2, and ASPC-1) were obtained from the Chinese Academy of Medical Science and Peking Union Medical College (Beijing, China), generously provided by Dr Hai HU. SUIT-2 was moderately differentiated, ASPC-1 was moderately to poorly differentiated, and PANC-1 was poorly differentiated[7]. All the cells were grown in a monolayer culture in a humidified atmosphere with 5% CO2 and 95% air at 37 °C. ASPC-1 and SUIT-2 cells were grown in RPMI-1640 (Sigma Chemical Co, St Louis, MO, USA) medium and PANC-1 cells in Dulbecco’s modified Eagle’s medium (Sigma Chemical Co, St Louis, MO, USA), respectively, supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin.
RNA isolation and RT-PCR The transcription levels of SHH, SMO, and EGFR in the pancreatic cancer cells were detected by RT-PCR analyses before and after the addition of cyclopamine and Iressa, alone or in combination. The pancreatic cancer cells were collected by centrifugation and the total RNA was isolated from the cultures using Trizol reagent (Qiagen, Hilden, Germany). After quantification, mRNA was transcribed into first-strand cDNA using the Avian Myeloblastosis Virus Reverse Transcriptase (AMV-RT) (Promega Biotech Co, Madison, WI, USA). Conventional PCR reactions were carried out in a total volume of 20 µL containing 1× PCR-buffer (Toyobo Co., Kita-ku, Osaka City, Japan), 150 µmol/L of each nucleotide, 20 pmol/L of each SHH, SMO, or EGFR primer, and 1 U of Taq polymerase (Toyobo, Co., Kita-ku, Osaka City, Japan). The PCR products were separated by agarose gel electrophoresis and visualized by ethidium bromide staining.
The following primer sequences were employed:
SHH (170 bp): sense 5'-GAAAGCAGAGAACTCGGTGG-3' and antisense 5'-GGAAAGTGAGGAAGTCGCTG)-3'; SMO (263 bp): sense 5'-ATCTCCACAGGAGAGACTGGTTCGG-3' and antisense 5'-AAAGTGGGCCTTGGGAACATG-3'; EGFR (194 bp): sense 5'-GTGGCTGGACTGCTCAAGAG-3' and antisense 5'-CTAGTCTCGAGTAGGCCTTTGTG-3'; β-actin (410 bp): sense 5'-CTACGAGCTGCCTGACG-3' and antisense 5'-AGAAGCATTTGCGGTGG-3'.
Western blot analysis The pancreatic cancer cells were solubilized in lysis buffer containing 50 mmol/L Tris-HCl, 1% Nonidet P-40 (NP-40), 0.25% Na-deoxycholate, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L phenylmethanesulfonyl fluoride(PMSF), and the protease inhibitors of the cocktail (Roche Diagnostics Ltd, Basel, Switzerland). The lysate were centrifuged at 10 000×g for 30 min to remove insoluble material. The total protein concentration was calculated according to the Bradford protein quantification method. Samples of equal amount were loaded onto a 10% SDS-PAGE gel and electrophoretically transferred to a Polyvinylidene Difluoride(PVDF) membrane. After blocking with 5% non-fat milk/ Tris-Tween buffer saline (TTBS), the membrane was incubated for 90 min with a highly specific primary antibody of anti-EGFR (sc-03, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-SHH (sc-1194, Santa Cruz Biotechno-logy, Santa Cruz, CA, USA), respectively, then washed with 0.05% Tween-20 in Tris buffer saline TBS and incubated with horseradish peroxidase-coupled secondary antibody for 60 min. Protein signals were detected using an electrochemilumi-nescence(ECL) reagent (Amersham Pharmacia Biotech, Buckingham, UK).
Iressa and cyclopamine treatment The logarithmically-growing pancreatic cancer cells were plated at a density of 1×104 cells/well into a 96-well plate, followed by 24 h of serum starvation to synchronize. Cyclopamine (Toronto Research Chemicals, Ontario, Canada) of different concentra-tions, alone or in combination with Iressa (Astra Zeneca Pharmaceuticals LP, Wilmington, DE and Cheshire, UK), were added to the culture medium. Iressa treatment was performed at 3 doses (1, 2.5, and 5 µmol/L, respectively), whereas cyclopamine treatment was performed at 2 dosages (2.5 and 5 µmol/L, respectively). DMSO was used at a final concentration of 0.1% in the control wells. After treatment for 72 h, the rate of cell growth and apoptosis was measured.
Methyl thiazolyl tetrazolium analysis 20 µL methyl thiazolyl tetrazolium (MTT) (5 g/L) was added to each well and incubated for an additional 4 h; the culture media were then discarded, followed by the addition of 0.15 mL DMSO, and then vibrated for 10 min. The absorbance was measured at 490 nm using a model 550 microplate reader. The inhibitory rates (IR) were calculated as follows: IR (%)=([1–absorbance of the treated wells]/[Absorbance of the control wells])×100.
Flow cytometry analysis The pancreatic cancer cell density was adjusted to (0.3–1.0)×107 cells/mL. The cells were serum starved for 24 h and then treated with different concentrations (2.5 or 5 µmol/L, respectively) of cyclopamine, alone or in combination with 1 µmol/L Iressa for 72 h. Then the cells were harvested with trypsin-EDTA to produce a single cell suspension. The cells were pelleted by centrifugation and washed twice with phosphate-buffered solution (PBS). Then the cell pellets were resuspended in 0.5 mL PBS and fixed in 5 mL ice-cold 70% ethanol at 4 °C. The fixed cells were spun down by centrifugation and the pellets were washed with PBS. After being resuspended in 1 mL PBS, the cells were incubated with RNase A (20 mg/L, Sigma Chemical Co, St Louis, MO, USA) and propidium iodide (PI) (50 mg/L, Sigma Chemical Co, St Louis, MO, USA) and shaken for 1 h at 37 °C in the dark. The stained cells were analyzed using a FACScan flow cytometer in combination with BD analysis II software (Becton Dickinson, Franklin Lakes, NJ, USA). The apoptosis rates were calculated.
Statistical analysis Data were expressed as mean±SD. The data were analyzed using one- or two-way ANOVA according to the experimental design applied. The simple and the repeated contrasts were applied to ANOVA in order to compare the various experiments with the control experiment as well as adjacent categories, respectively. All statistical analyses were performed using the SPSS 11.0 software package for Windows (SPSS, Chicago, IL, USA). A two-tailed P-value less than 0.05 was considered statistically significant.
Results
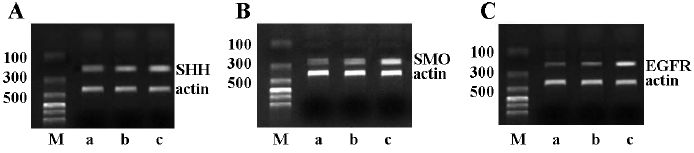
mRNA transcription levels of SHH, SMO, and EGFR SHH, SMO and EGFR mRNA were transcribed in all of the 3 pancreatic cancer cell lines. The PANC-1 cells exhibited weak transcription, whereas the other 2 cell lines were found to have a high transcription level of SHH, SMO, and EGFR at the mRNA level (Figure 1).
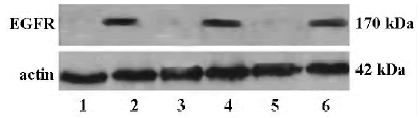
Detection of SHH and EGFR protein expressions by Western blot analysis The supernatant of the cultured cells in the 3 groups was collected and analyzed by Western blotting. The SHH and EGFR protein expressions were found in all the 3 cell lines, and the expression level of the SHH and EGFR proteins were lower in PANC-1 than in ASPC-1 and SUIT-2 (Figure 2).

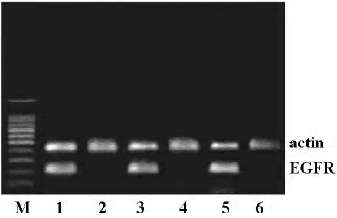
Effect of cyclopamine on the expression of EGFR To identify the relationship between the SHH and EGFR signaling pathway, the effect of cyclopamine on the expression of EGFR was investigated. After exposure to cyclopamine (5 µmol/L) for 48 h, RT-PCR and Western blot analysis were carried out and revealed that cyclopamine could significantly suppress the transcription and expression levels of EGFR (Figures 3, 4).
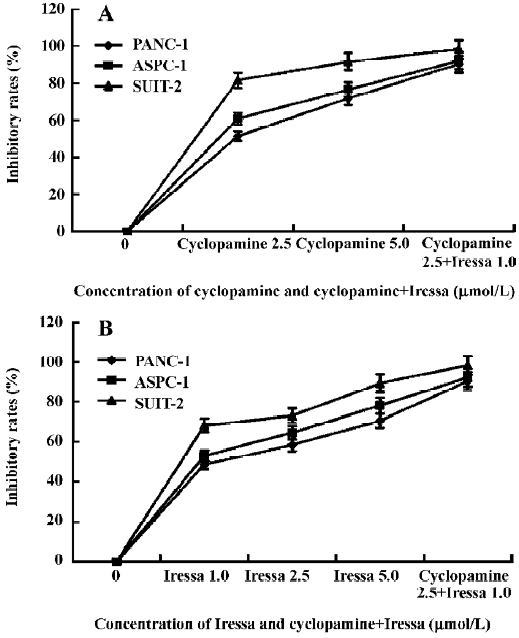
Effect of cyclopamine and Iressa on cell proliferation A MTT assay was used to investigate the cell viability of PANC-1, ASPC-1, and SUIT-2 after cyclopamine and Iressa treatment. As shown in Figure 5, cyclopamine could inhibit the growth of the pancreatic cancer cells in a dose-dependent manner. SUIT-2 cells demonstrated the strongest response among the 3 cell lines due to the overexpression of SHH signaling. Even at a minimum dose of 2.5 µmol/L cyclopamine, SUIT-2 cells showed a significant (P<0.001) growth reduction (81.6%), whereas PANC-1 and ASPC-1 cells showed a moderate response (51.4% and P=0.047 and 60.8% and P=0.035, respectively). At the dose of 5 µmol/L, almost complete growth inhibition (91.7% reduction) was observed in SUIT-2 cells. At this dose, PANC-1 and ASPC-1 also showed significant reduction in growth (71.9% and P=0.032 and 76.6% and P=0.016, respectively). The EGFR tyrosine kinase inhibitor Iressa also induced a growth inhibitory effect in a dose-dependent manner. The most significant impact on growth inhibition was observed in SUIT-2 cells, followed by ASPC-1, whereas PANC-1 demonstrated a moderate response. At the dose of 5 µmol/L, almost complete (89.3%) growth reduction was observed in SUIT-2 cells, a 78.2% reduction was observed in ASPC-1 cells, and a 70.6% reduction was seen in PANC-1 cells (P<0.01), respectively. More-over, the combined use of 2.5 µmol/L cyclopamine and 1 µmol/L Iressa induced an enhanced inhibitory effect, which was much more than that of 5 µmol/L cyclopamine or 5 µmol/L Iressa alone.
Effects of cyclopamine and Iressa on cell cycle progression In order to examine the effects of cyclopamine and/or Iressa on cell cycle progression, the pancreatic cancer cells were treated with the indicated concentrations of cyclopamine (2.5 and 5 µmol/L, respectively) and Iressa (1, 2.5, and 5 µmol/L, respectively), alone or in combination for 72 h. The percentage of cells in each phase of the cell cycle, G0/G1, S, and G2/M was determined by flow cytometry after the cells were treated. As shown in Table 1, the percentage of the cell population of the G0/G1 phase was significantly increased, while that in S and G2/M phases decreased. Cyclopamine and/or Iressa caused cell cycle arrest in the G0/G1 phase in a dose-dependent manner.
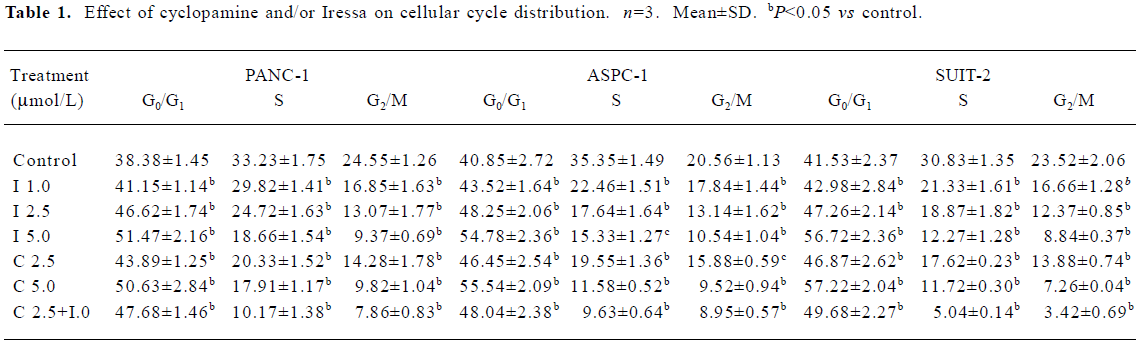
Full table
Effect of cyclopamine and Iressa on apoptosis After the pancreatic cancer cells were exposed to the indicated concentrations of cyclopamine (2.5 µmol/L) and Iressa (1.0 µmol/L), alone or in combination for 72 h, a flow cytometry analysis was carried out and revealed that the apoptotic rate of PANC-1 was 18.17%±0.73%, 11.82%±0.24%, and 32.55%± 0.56%, respectively. The apoptotic rate in the control group was 1.38%± 0.45% and the apoptotic rate of SUIT-2 was higher than the other 2 cell lines. Cyclopamine and Iressa induced apoptosis of the pancreatic cancer cells in a dose-dependent manner. Moreover, 2.5 µmol/L cyclopamine plus 1 µmol/L Iressa was more effective to all tested pancreatic cancer cells than agents alone and this combination caused the death of the majority of cells (Table 2).
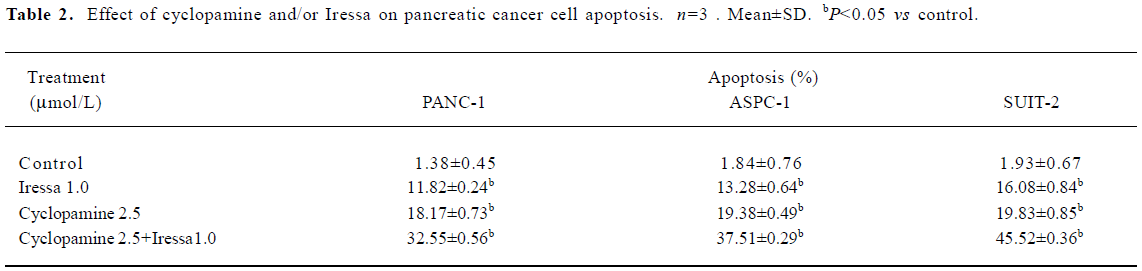
Full table
Discussion
SHH signaling is important for normal axial patterning of the mammalian embryo and has been proved to be essential for foregut development. Recently, some studies have explored the role of inappropriate activation of the SHH signaling pathway in several types of gastrointestinal tumors[5]. On the other hand, EGFR could promote proliferation and differentiation of some epithelial cells both in vitro and in vivo[8]. EGFR has been involved in the development of many tumors, such as lung, breast, prostate, and colon cancers[9]. In our study, the overexpression of endogenous SHH ligands, SMO, and EGFR were detected by RT-PCR and Western blot analysis in pancreatic cancer cell lines. However, the expression level of these molecules was different in these cells. SUIT-2 cells have the highest expression level, whereas PANC-1 cells have the lowest. The concomitant elevated expression of SHH and EGFR appears to be implicated in the activation of mitotic signaling[10,11], resulting in a continuance proliferation of pancreatic cancer cells.
In our studies, when the pancreatic cancer cells were treated with cyclopamine, the mRNA and protein levels of EGFR decreased accordingly, which means that the cyclopamine treatment of pancreatic cancer cells could downregulate the expression of EGFR. It has been reported that the SHH ligand could induce the growth of pancreatic cancer cells in an autocrine fashion[12]. The stimulation of the SHH ligand has been reported to increase Drosophila EGFR (DER) signaling by upregulating the expression of the EGFR[13]. Furthermore, it has been reported that the aberrant activation of SHH signaling in the precursor cells results in the initiation of some tumors, such as brain and skin[14,15]. Although it remains uncertain as to whether or not these tumors come from the aberrant activation of SHH and EGFR in stem cells or differentiated cells, it seems that there must be some interaction between these signaling pathways[10,16,17].
As an inhibitor of the SHH signaling pathway, cyclopamine is an alkaloid isolated from the Veratrum californicum plant[18]. Berman et al found that treatment with cyclopamine inhibited the growth of digestive tract cancer significantly[5]. The inhibitory role of cyclopamine is being tested for other cancers as well, for example medulloblastoma[19], breast[20], and prostate cancers[10,21,22]. In our studies, it was demonstrated that cyclopamine could induce the inhibition in pancreatic cancer cells with activated SHH signaling, but different cell lines present variable reactions to cyclopamine. For SUIT-2 cells, treatment with 2.5 µmol/L cyclopamine induced an inhibitory rate of 81.6%, whereas the rate of PANC-1 and ASPC-1 cells was 51.4% and 60.8%, respectively. This difference might be due to the different expression of SHH in different cell lines. Perhaps other genetic alterations, such as the Indian Hedgehog (IHH) and EGFR signaling pathways, may contribute to it. Recently, a report indicated that IHH, another member of the HH family, may be the dominant ligand expressed in both pancreatic cancer and chronic pancreatitis[23]. Further understanding of the molecular basis for cell sensitivity to cyclopamine will help us design better ways to manipulate pancreatic cancer in the future. Thus, it may be possible in the future to treat the subsets of pancreatic cancer with SHH signaling inhibitors.
It has been reported that the blockade of the EGFR signaling pathway with Iressa, a selective EGFR inhibitor, resulted in antiproliferative activity in human cancer cell lines of different histological types, such as ovarian, breast, and colon cancers[24]. Our results indicated that Iressa also inhibited the proliferation of pancreatic cancer cells in a dose-dependent manner. Moreover, the combination of 2.5 µmol/L cyclopamine and 1.0 µmol/L Iressa resulted in a higher inhibitory effect in pancreatic cancer cells than any single agent. Hence, these pathways might act cooperatively for the proliferation of pancreatic cancer cells. In our study, the pancreatic cancer cells were arrested in the G0/G1 phase after cyclopamine treatment, concomitant with a decrease of the cell number in the S and G2/M phases. It has been reported that the SHH signaling could be activated by the upregulation of cell cycle regulators, such as cyclins[25]. Therefore, cyclopamine could downregulate the expression of cell cycle regulators through the inhibition of SHH signaling, and as a result, the mitogenic effect induced by other growth factors, such as EGF, might be balanced out[10].
In this study, the apoptotic effect induced by cyclopamine, alone or at lower concentrations with Iressa in pancreatic cancer cells was also investigated. Cyclopamine and Iressa induced apoptosis of pancreatic cancer cells in a dose-dependent manner. Moreover, 2.5 µmol/L cyclopamine plus 1 µmol/L Iressa caused a stronger inhibitory effect to all tested pancreatic cancer cells than agents alone and this combination caused the death of the majority of cells.
Further studies of SHH signaling in different stages of pancreatic cancers, especially at early stages, will facilitate early diagnosis of pancreatic cancer through the detection of SHH signaling. Moreover, the simultaneous blockade of the SHH and EGFR signaling pathways leads to an arrest of growth and an increase of apoptosis in pancreatic cancer cells. These pathways may offer an exciting, new therapeutic approach for pancreatic carcinoma which is still treated by palliative procedures or highly toxic drugs. The selective inhibition of signaling pathways activated in precancerous cells should be possible without disturbing other pathways necessary for normal cell function, also minimizing toxicity from such therapy. Cyclopamine appeared to selectively induce apoptosis in tumor cells without adverse effects on normal tissues in vivo[26], thus, it was an ideal drug with potential to prevent the progression of pancreatic cancer cells.
References
- Moon HJ, An JY, Heo JS, Choi SH, Joh JW, Kim YI. Predicting survival after surgical resection for pancreatic ductal adenocarci-noma. Pancreas 2006;32:37-43.
- Thayer SP, Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003;425:851-6.
- Hebrok M, Kim SK, St Jacques B, McMahon AP, Melton DA. Regulation of pancreas development by hedgehog signaling. Development 2000;127:4905-13.
- Fu JR, Liu WL, Zhou JF, Sun HY, Xu HZ, Luo L, et al. Sonic hedgehog protein promotes bone marrow-derived endothelial progenitor cell proliferation, migration and VEGF production via PI 3-kinase/Akt signaling pathways. Acta Pharmacol Sin 2006;27:685-93.
- Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003;425:846-51.
- Wang Z, Sengupta R, Banerjee S, Li Y, Zhang Y, Rahman KMW, et al. Epidermal growth factor receptor-related protein inhibits cell growth and invasion in pancreatic cancer. Cancer Res 2006;66:7653-60.
- Sipos B, Moser S, Kalthoff H, Torok V, Lohr M, Kloppel G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: towards the establishment of an in vitro research platform. Virchows Arch 2003;442:444-52.
- Carpenter G, Cohen S. Epidermal growth factor. J Biol Chem 1990;265:7709-12.
- Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995;19:183-232.
- Mimeault M, Moore E, Moniaux N, Hénichart JP, Depreux P, Lin MF, et al. Cytotoxic effects induced by a combination of cyclopamine and gefitinib, the selective hedgehog and epidermal growth factor receptor signaling inhibitors, in prostate cancer cells. Int J Cancer 2006;118:1022-31.
- Sui G, Bonde P, Dhara S, Broor A, Wang J, Marti G, et al. Epidermal growth factor receptor and hedgehog signaling pathways are active in esophageal cancer cells from rat reflux model. J Surg Res 2006;134:1-9.
- Kayed H, Kleeff J, Osman T, Keleg S, Buchler MW, Friess H. Hedgehog signaling in the normal and diseased pancreas. Pancreas 2006;32:119-29.
- Amin A, Li Y, Finkelstein R. Hedgehog activates the EGF receptor pathway during Drosophila head development. Development 1999;126:2623-30.
- Dahmane N, Sanchez P, Gitton Y, Palma V, Sun T, Beyna M, et al. The Sonic Hedgehog–Gli pathway regulates dorsal brain growth and tumorigenesis. Development 2001;128:5201-12.
- Altaba A, Sanchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer 2002;2:361-72.
- Wessells RJ, Grumbling G, Donaldson T, Wang SY, Simcox A. Tissue-specific regulation of vein/EGF receptor signaling in Drosophila. Dev Biol 1999;216:243-59.
- Altaba A, Stecca B, Sanchez P. Hedgehog-Gli signaling in brain tumors: stem cells and paradevelopmental programs in cancer. Cancer Lett 2004;204:145-57.
- Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998;125:3553-62.
- Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002;297:1559-61.
- Kubo M, Nakamura M, Tasaki A, Yamanaka N, Nakashima H, Nomura M, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res 2004;64:6071-4.
- Sanchez P, Hernandez AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A, et al. Inhibition of prostate cancer proliferation by interference with sonic hedgehog-gli1 signaling. Proc Natl Acad Sci USA 2004;101:12561-6.
- Karhadkar SS, Steven Bova G, Abdallah N, Dhara S, Gardner D, Maitra A, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004;431:707-12.
- Kayed H, Kleeff J, Keleg S, Guo J, Ketterer K, Berberat PO, et al. Indian hedgehog signaling pathway: expression and regulation in pancreatic cancer. Int J Cancer 2004;110:668-76.
- Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res 2000;6:2053-63.
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol 2000;20:9055-67.
- Qualtrough D, Buda A, Gaffield W, Williams AC, Paraskeva C. Hedgehog signalling in colorectal tumour cells: induction of apoptosis with cyclopamine treatment. Int J Cancer 2004;110:831-7.