
Antitumor effects of Chi-Shen extract from Salvia miltiorrhiza and Paeoniae radix on human hepatocellular carcinoma cells
Introduction
Human hepatoma is one of the most frequent cancers in the world and it has the characteristics of secret coming on, including rapid growth rate, strong malignancy, easy invasion, metastasis and bad prognosis, and the occurring of hepatoma presented an ascending trend. However, the curative effect of current therapies in liver cancer is not perfect[1]. Treatment is difficult if the cancer has spread beyond the liver. Therefore, it is very important to find new medicine to treat hepatoma.
Salvia miltiorrhiza has been used as a traditional medicine in China for centuries. It was commonly used for the treatment of cardiovascular diseases, such as angina pectoris, myocardial infarction, and stroke[2]. Some studies also indicated that the extract of Salvia miltiorrhiza had antitumor potential[3]. The aqueous extract of Salvia miltiorrhiza could inhibit cell growth and induce apoptosis in human hepatocellular carcinoma cell line HepG2 cells[4] and salvianolic acid B, a water-soluble compound from Salvia miltiorrhiza that has anti-oxidative and free radical scavenging effects[5]. It was recently reported that it could effectively inhibit the proliferation of the chronic myelogenous leukemia K562 cell line[6]. However, the underlying anticancer mechanisms of salvianolic acid B and the aqueous extract of Salvia miltiorrhiza are unclear.
Paeoniae radix is the root of Paeonia lactiflora Pallas and is a crude drug used in many traditional prescriptions in China and Japan[7]. Paeoniae radix is a potential anti-aging and anticarcinogenesis agent as it has been reported to inhibit oxidative DNA cleavage induced by various oxidative DNA damage chemicals[8]. Paeoniflorin, a water-soluble compound from Paeoniae radix, could induce apoptosis of lymphocytes[9]. Lee et al showed that the water soluble extract of Paeoniae radix (PRE) had an inhibitory effect on the growth of both the HepG2 and Hep3B cell lines. The induction of internucleosomal DNA fragmentation and chromatin condensation appearance, and the accumulation of the sub-G1 phase of cell cycle profile in PRE-treated hepatoma cells showed that the cytotoxicity of PRE to hepatoma cells was through the activation of apoptosis in a p53-independent pathway. The underlying mechanism of PRE as an anticancer agent has not been completely defined[10].
Compounds that block or suppress the proliferation of tumor cells by inducing apoptosis are considered to have potential as antitumor agents[11]. In this study, Chi-Shen extracts (CSE) were composed of the water-soluble extracts of Salvia miltiorrhiza, mainly containing salvianolic acid B and the glycoside extract of Paeoniae radix containing paeoniflorin at a ratio of 7:3. The objective of this study was to examine the in vitro antitumor activities of CSE on hepatoma cell line HepG2, which was through the activation of apoptosis, evidenced by the induction of internucleosomal DNA fragmentation and the accumulation of the sub-G1 phase of cell cycle profile. The activation of apoptosis by CSE was dependent on the caspase pathways and probably through altering the expression level of the Bcl-2 family and p53 as identified by Western blotting analysis.
Materials and methods
Materials Slices of Salvia miltiorrhiza and Paeoniae radix were purchased at a local market in Wuhan, a well-known production area for Salvia miltiorrhiza and Paeoniae radix in China. Methyl thiazolyl tetrazolium (MTT), acridine orange (AO), ethidium bromide (EB), RNase A, propidium iodide (PI), and trypsin were purchased from Sigma Chemical Co (St Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), low melting point agarose, and normal melting point agarose were from GIBCO (Grand Island, NY, USA). The apoptosis ladder detection kit was from Wako Pure Chemical Industries (Osaka, Japan). AMV reverse transcriptase was purchased from Promega (Madison, WI, USA). The caspase colorimetric assay kit was obtained from BioVision (Mountain View, CA, USA). The micro BCA protein assay kit was from Beijing Biosea Biotechnical Corporation (Beijing, China). RNAzol was from Omega Biotek (Doraville, GA, USA). Bcl-2, Bax, p53, and β-actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Other chemicals and reagents used were obtained from Sigma (USA). All chemicals and reagents were of analytical grade.
Preparation of CSE extracts The dried root slices of Salvia miltiorrhiza were percolated with 30% ethanol at room temperature. After evaporation of the solvent, the concentrated aqueous extract was acidified with hydrochloric acid (pH 2–3) and then extracted with acetic ether. The organic phase was then concentrated and dried to yield the water-soluble extract of Salvia miltiorrhiza, which mainly contained salvianolic acid B. The crude powder of Paeoniae radix was extracted with 50% ethanol and then filtered. After evaporation of the solvent, the crude extract was dissolved in water and applied to a macroporous resin column that was washed with water and 30% ethanol successively. The eluent was then concentrated, dried to yield the glycoside extract of Paeoniae radix, which mainly contained paeoniflorin. CSE, which was composed of the water-soluble extract of Salvia miltiorrhiza and the glycoside extract of Paeoniae radix, at a ratio of 7:3, was dissolved in phosphate-buffered saline (PBS) for storage at 4 ℃ and used in all subsequent experi-ments. The final storage concentration was 100 mg/mL, and the concentration used in the experiment was based on the dry weight of the extract (mg/mL).
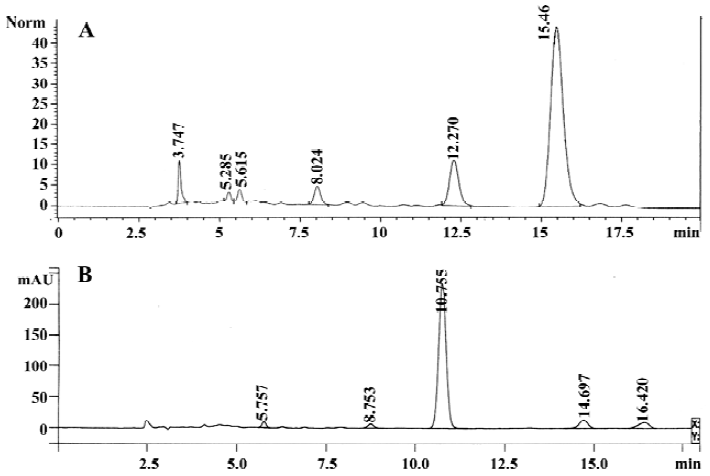
HPLC assay The extract of Salvia miltiorrhiza, which mainly contained salvianolic acid B used for HPLC analysis, was performed in Agilent 1100 high-performance liquid chromatograph (Agilent, Waldbronn, Germany), with a Kromasil C18 column (Hichrom, Reading, UK; 5 µm, 4.6 mm×250 mm), eluted with the mobile phase containing formic acid-0.2% H3PO4 (17:83), at a flow rate of 1.0 mL/min and a detector wavelength of 280 nm. The whole run time was 20 min, while the retention time of salvianolic acid B was 15.460 min (Figure 1A). The extract of Paeoniae radix mainly containing paeoniflorin used for the HPLC analysis was performed in a Varian Prostar 210 high-performance liquid chromatograph (Palo Alto, CA, USA), with a Zorbax Eclipse XDB-C18 column (5 µm, 4.6×250 mm, Agilent, CA, USA), eluted with the mobile phase containing acetonitrile-H2O (17:83), at a flow rate of 1.0 mL/min and a detector wavelength of 230 nm. The whole run time was 20 min, while the retention time of paeoniflorin was 10.755 min (Figure 1B).
Cell culture The human hepatoma cell line (HepG2) was obtained from the China Center for Type Culture Collection (Wuhan, China). The cells were cultured with DMEM with 10% (v/v) heat-inactivated FBS, 100 µg/mL streptomycin, and 100 unit/mL penicillin in 100 mL culture flasks in a humidified atmosphere at 37 ℃ with 5% CO2. Single cell detachment was achieved with incubation at 37 ℃ with trypsin-EDTA.
Cell viability assay Cell viability was measured by 3-(4,5)-2,5-diphenyltetrazolium bromide (MTT) assay. In brief, the HepG2 cells were treated with CSE at concentrations of 0.25, 0.5, 1.0, and 2.0 mg/mL for 12, 18, 24, and 36 h, respec-tively, and the control group was treated with the same amount of medium. 5-Fluorouracil (5-FU), an anticancer drug inhibiting DNA and RNA synthesis, was used as a standard control. After the MTT labeling reagent was added and incubated for 4 h at 37 ℃, they were incubated for 12 h with the 10% sodium dodecyl sulphate (SDS) solution. The absorbance (at a test wavelength of 540 nm and a reference wavelength of 690 nm) was measured by a microtiter plate reader (Molecular Devices, Silicon Valley, CA, USA). The optical density (OD) was calculated as the difference between the absorbance from the reference and test wave-lengths. The percentage of viability was calculated as the following formula: (viable cells) %=(OD of drug-treated sample/OD of untreated sample)×100.
Morphological examination The cells were harvested and washed 3 times with PBS after being incubated with different concentrations of CSE for 12, 18, 24, and 36 h respectively, and were stained with 10 µg/mL AO/EB for 5 min. Apoptotic morphology was observed by a fluorescence microscope (BX51, Olympus, Shinjuku-ku, Tokyo, Japan) after AO/EB staining.
Analysis of DNA fragmentation The cells were harvested and washed 3 times with PBS after being incubated with different concentrations of CSE for 24 h. DNA was extracted with the apoptosis ladder detection kit, analyzed by 1.0% agarose gel electrophoresis, and then stained with EB.
DNA content and cell cycle analyzed by flow cytometry The DNA content and the cell cycle of HepG2 cells were determined by flow cytometry. Briefly, at the end of the various designated treatments, the cells were scrapped, washed, and fixed with 80% ice-cold ethanol at -20 ℃ overnight. The cells were incubated with freshly prepared PI staining buffer containing 0.25 mg/mL RNase A, 2 mmol/mL EDTA, and 0.1 mg/mL PI in PBS. After incubation in dark conditions at 37 ℃ for 30 min, the fluorescence of 10 000 cells was analyzed using a flow cytometer with an argon ion laser (488 nm) as the excitation source and Cell Quest software (Becton Dickinson, San Jose, CA, USA). The percentage of degraded DNA was determined by the number of hypodiploid DNA cells, and the change of cell cycle was determined at the same time.
Caspase activity assay Caspase activation was measured using a caspase colorimetric assay kit as described by the manufacturer. Briefly, after being treated with 1.0 mg/mL CSE for the indicated periods of time, the cells were harvested, pelleted, and frozen on dry ice. Cell lysis buffer was added to the cell pellets, and protein concentration was determined by a micro BCA kit. Then, 100 µg protein was suspended in 50 µL cell lysis buffer for each assay, and 50 µL of 2× reaction buffer with 10 mmol/L dithiothreitol (DTT) was added and incubated at 4 ℃. The substrates of Ile–Glu–Thr–Asp conjugated to p-nitroanilide (IETD–pNA), Leu–Glu–His–Asp conjugated to p-NA (LEHD–pNA), and Asp–Glu–Val–Asp conjugated to p-NA (DEVD–pNA) were added into the tubes, respectively. Formations of p-NA were measured by an ELISA micro-plate reader (Thermo, Vantaa, Finland) at a wavelength of 405 nm after the samples were incubated for 1.5 h at 37 ℃. The activation of caspases-8, -9, and -3 were analyzed in parallel.
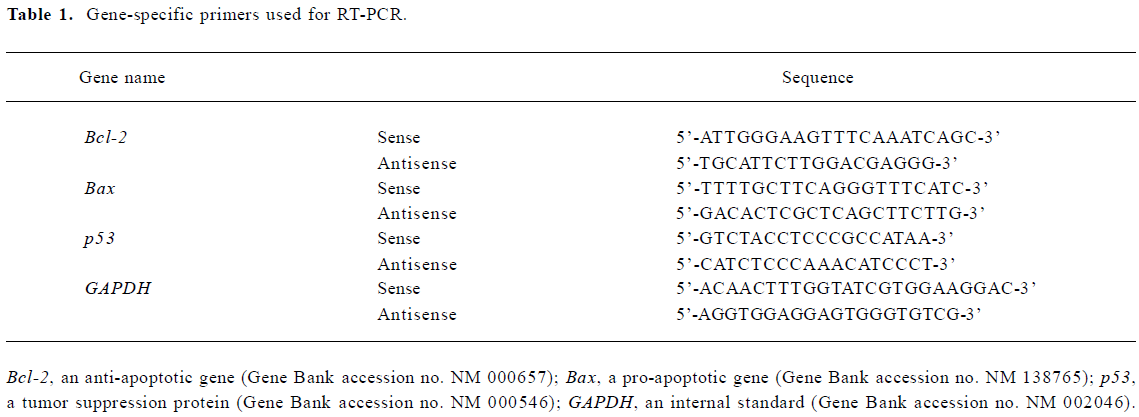
RNA purification and RT-PCR The expression of apoptosis-related genes, such as Bcl-2, Bax, and p53 were determined by RT-PCR. Total RNA was isolated with RNAzol according to the manufacturer’s instruction. Single-strand cDNA was synthesized from 2 µg total RNA using AMV reverse transcriptase. Bcl-2, Bax, p53, and GAPDH (an internal standard) cDNA were amplified by PCR with specific primers (Table 1). The annealing temperature was 58 ℃ for Bcl-2, 55 ℃ for Bax, 55 ℃ for p53, and 58 ℃ for GAPDH. The amplified fragment sizes of Bcl-2, Bax, p53, and GAPDH were 301, 114, 316, and 387 bp, respectively. The RT-PCR products were electrophoresed on the 1.0% agarose gel and visualized by staining with EB.
Full table
Western blotting For Western blotting, the cells were first treated for 24 h with different concentrations of CSE, 2×106 cells were washed twice with ice-cold PBS, lysed for 30 min at 4 ℃, and then the debris was removed by centrifugation for 15 min at 12 000×g at 4 ℃. The equivalent amount of protein (20 µg) were separated by 10% SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were first stained to confirm uniform transfer of all samples and then incubated in blocking solution for 2 h at room temperature. The filters were hybridized first with monoclonal antibody (anti-Bcl-2, anti-Bax and anti-p53, respectively) at a dilution of 1:1000 for 2 h, followed by extensive washes with PBS twice and TBST (Tris Buffered Saline supplemented with 0.1% Tween-20) twice. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibody at a dilution of 1:1000 for 1 h, and washed with TBST. As a loading control, β-actin was also detected. The immunoreactive proteins were detected using an ECL Western blotting detection system (Beyotime Institute of Biotechnology, Haimen, China).
Statistical analysis Data are presented as mean±SD. The differences among different groups were analyzed using one-way ANOVA with Scheffe’s test. A P-value less than 0.05 was considered statistically significant.
Results
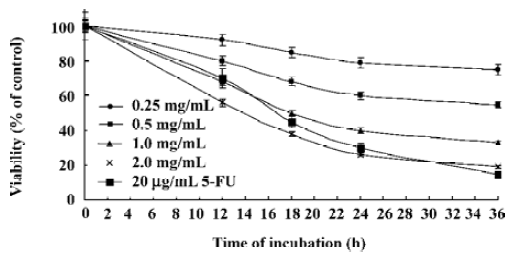
Effect of CSE on viability in HepG2 cells The potential effect of CSE was investigated on the viability of a human hepatoma cell line (HepG2). MTT assay was performed to evaluate the cell viabilities (Figure 2). Compared to the untreated cells (taken as 100% viable), different concentrations of CSE or 5-FU showed a time- and dose-dependent inhibition of HepG2 cell proliferation. No significant decrease in cell viability was observed when HepG2 cells were treated with 0.25 mg/mL CSE for 36 h. However, exposure of HepG2 cells to CSE at 1.0 and 2.0 mg/mL caused a marked decrease in overall viability from 100% to 33% and 19% of untreated control levels, respectively.
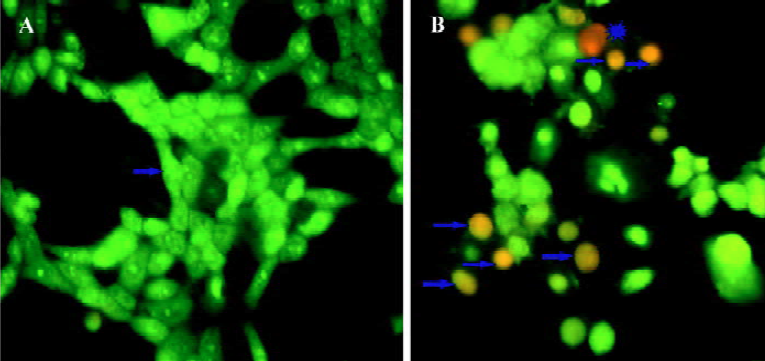
CSE treatment caused apoptosis To determine whether the cell growth arrest caused by CSE treatment was related to the induction of apoptosis, morphological observation with AO/EB staining was detected by fluorescence micro-scopy. The HepG2 cells treated with 1.0 mg/mL CSE for 24 h were stained and observed. Green live cells with normal morphology were seen in the control group (Figure 3A), while early apoptotic cells with nuclear margination and chromatin condensation, and later apoptotic cells with fragmented chromatin, were orange in the experimental group treated with 1.0 mg/mL CSE (Figure 3B). The results suggested that CSE induced marked apoptotic morphology in HepG2 cells.
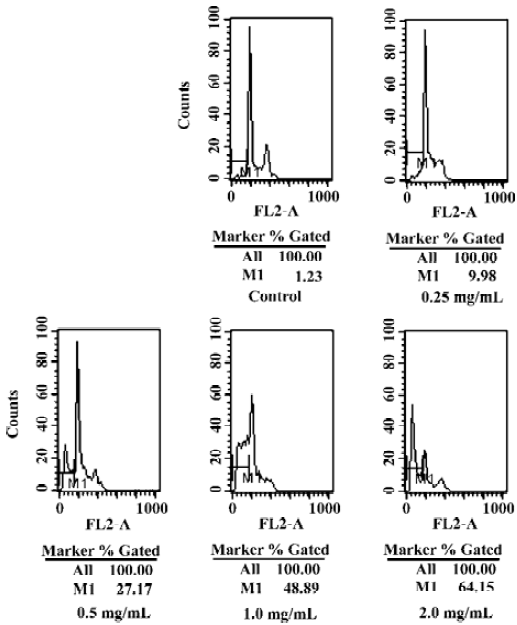
The effect of CSE on the intranuclear DNA fragmentation was further examined. After CSE treatment (1.0 and 2.0 mg/mL) for 24 h, DNA fragmentation was observed (Figure 4); no DNA ladder appeared in the control group. Apoptosis was further quantified by flow cytometry. Based on a previous report, cells with sub-G1 DNA content were scored as apoptosis. Apoptosis could result in the progressive generation of particles corresponding to hypodiploid DNA content, which reflects DNA fragmentation[12]. In our study, CSE treatment resulted in an increment of the sub-G1 fraction (M1 fraction of Figure 5) in HepG2 cells, which indicated that apoptosis induced by CSE treatment was in a dose-dependent manner. These results were in accordance with those obtained from the DNA fragmentation assay.

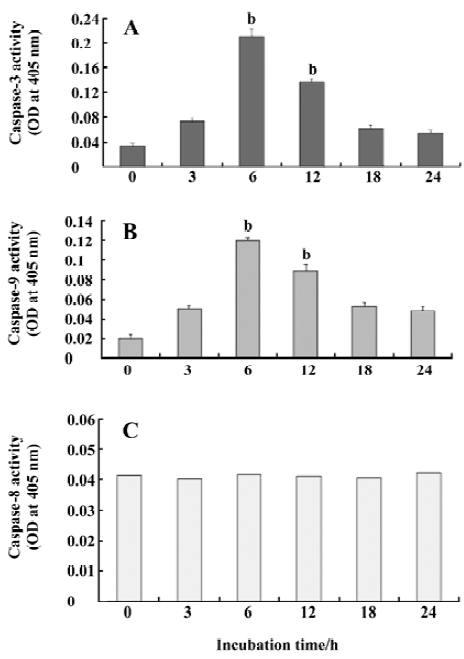
Activation of caspase by CSE To identify whether caspases are involved in the mechanism of apoptosis, we measured the catalytic activity of caspases-3, -8, and -9. The results showed that caspases-3 and -9 were activated and peaked at 6 h of CSE treatment (Figure 6A,6B). However, the results indicated that activated caspase-8 did not change significantly (Figure 6C).
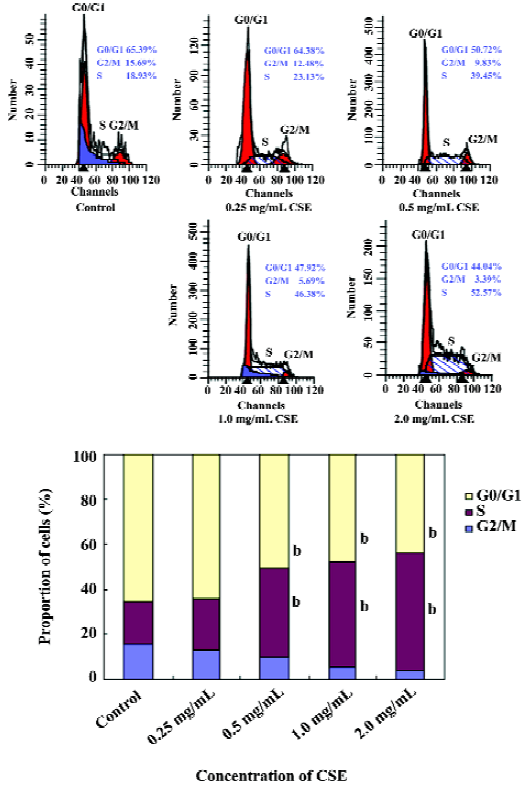
Effect of CSE on cell cycle distribution To test whether there was an induction of cell growth arrest of CSE treatment in HepG2 cells, cell cycle analyses were performed. HepG2 cells treated with the rising CSE concentration series (0.25–2.0 mg/mL) for 24 h had a dose-dependent growth arrest, as did the cells in the S phase. The cell proportion which decreased in the G2/M phase (Figure 7). The S-phase arrest of HepG2 cells was significant at above 1 mg/mL of CSE. These data indicated that CSE attracted cell cycle to the S phase in HepG2 cells.
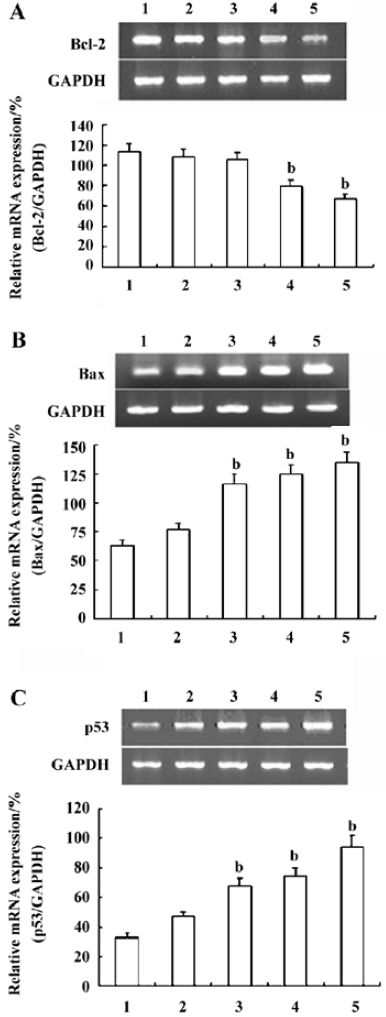
Effects of CSE on the transcription of Bcl-2, Bax, and p53 in HepG2 cells The transcription of Bcl-2, Bax, and p53 was detected by RT-PCR analysis. The results revealed that the transcription of Bcl-2 was downregulated, while that of Bax and p53 was considerably upregulated when the HepG2 cells were treated with CSE for 24 h (CSE over 0.25 mg/mL) in dose-dependent manner (Figure 8). This suggested that the downregulation of Bcl-2 and the upregulation of Bax and p53 may play an important role in CSE-induced apoptosis in HepG2 cells.
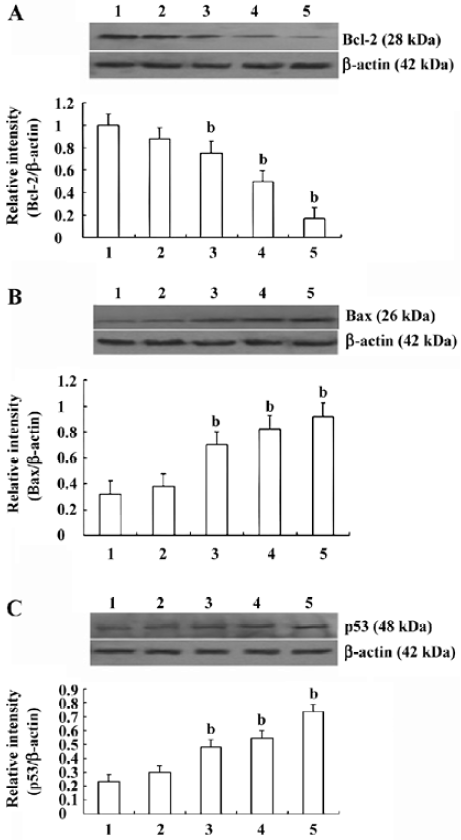
Effects of CSE on the expression of Bcl-2, Bax, and p53 in HepG2 cells The results of the Western blot analyses of Bcl-2, Bax, and p53 in HepG2 cells with or without CSE are shown in Figure 9. CSE stimulated the expression of Bax and p53 in a dose-dependent manner (Figure 9B, 9C). The expression of Bcl-2, an anti-apoptotic protein, decreased in a dose-dependent manner (Figure 9A).
Discussion
In a previous study, we verified that the antifibrotic and antitumor activities of CSE were stronger than that of single extract. The best proportion of the water-soluble extract of Salvia miltiorrhiza and the glycoside extract of Paeoniae radix was at a ratio of 7:3. The CSE capsule for relieving hepatic fibrosis has been applied in clinical studies in China. In the present study, the main aim of the investigation was to explore the antihepatoma potential of CSE and its mechanisms. Many investigations have been carried out worldwide to discover naturally-occurring compounds which can suppress or prevent the process of carcinogenesis[10,13]. Natural compounds fit into a mechanism-based approach that targets whole pathways and sets of intracellular events rather than a single enzyme, as do many synthetic drugs. This offers a less specific, but perhaps more effective strategy, for cancer therapy by inducing the combination of effects that may counteract the metabolic alterations related to cancer promotion[14]. For example, a capsicum and green tea mixture has been reported to exhibit 100 times greater potency with respect to anticancer activity in a number of cell lines, than that of green tea alone on a weight basis[15]. Early research reported that the extracts of Salvia miltiorrhiza and Paeoniae radix both have been documented to posses antitumor potential[6,10]. Therefore, in this study, CSE was mainly composed of salvianolic acid B and paeoniflorin, which are water-soluble extracts from Salvia miltiorrhiza and Paeoniae radix, respectively. Here, we showed that CSE was able to inhibit the growth of the human hepatoma cell line (HepG2) in a dose-dependent manner, which exhibited a similar potency as the commercial anticancer agent 5-FU (Figure 2).
Hepatocellular carcinomas (HCC) with poor prognosis are characterized by rapid cell proliferation and strong expression of anti-apoptotic genes[16], which suggests that they are mainly due to incomplete cell cycle arrest and apoptosis resistance under conventional therapies[17]. Recent data have shown that apoptosis, especially the caspase-mediated cell death, plays an important role in the etiology, pathogenesis, and therapy of a variety of human malignancies, such as human HCC, and the cytotoxic effects of many antihepatocellular carcinoma drugs are based on the induction of apoptosis[18]. Agents that can induce apoptosis in cells are considered to be potentially useful for the management and treatment of cancer[13]. Apoptosis is characterized by cell shrinkage, chromatin condensation, DNA fragmentation, sub-G1 DNA peak, and the activation of specific cysteine proteases known as caspases[12]. In the present study, DNA fragmentation with a ladder pattern characteristic of apoptosis was observed in HepG2 cells treated with CSE (1.0 and 2.0 mg/mL) for 24 h (Figure 4). We found that the growth inhibitory activity of CSE was associated with the induction of apoptosis in HepG2 cells. Data from an apoptosis assay showed that CSE induced obvious apoptosis in hepatoma HepG2 cells, presenting a dose-dependent manner of apoptosis-specific morphological changes (Figure 3) and sub-G1 peak (Figure 5). A noticeable phenomenon was that the apoptotic rate contrarily increased to 64.15% after treatment with 2.0 mg/ml CSE (Figure 5). This showed that CSE inhibited the proliferation of HepG2 cells through inducing cell apoptosis.
We investigated the effect of CSE on the levels of 3 members of the Bcl-2 family, the pro-apoptotic Bax and the anti-apoptotic Bcl-2, which regulate mitochondrial apoptosis[19]. When Bax was overexpressed in cells, apoptotic death in response to death signals was accelerated, earning its designation as a death agonist. When Bcl-2 was overexpressed, it heterodimerized with Bax and death was repressed, thus the ratio of Bcl-2 to Bax is important in determining susceptibility to apoptosis[20]. Following the exposure to CSE, we found an increase of Bax mRNA and protein level (Figures 8A, 9A) in the HepG2 cells, paralleled by a downregulation of Bcl-2 mRNA and protein level (Figures 8B, Figure 9B). This indicates that the mitochondria is an important target for CSE actions. These findings were similar to data obtained with hepatoma carcinoma cells exposed to gefitinib as pro-apoptotic agent[21]. Furthermore, caspases are a ubiquitous family of cysteine proteases[22]. DNA fragmentation, a hallmark of apoptosis, can be achieved, at least in part, through the intrinsic and extrinsic pathways. The activation of caspases-9 and -3 is the result of the induction of the intrinsic pathway, while in the extrinsic pathway, caspase-8 and then caspase-3 were activated[23]. In both pathways, the initiator caspase cleaves and activates downstream effector caspases, such as caspase-3[12]. It could be concluded that CSE induces apoptosis in HepG2 cells through the intrinsic pathway of the caspase cascade as only caspases-9 and -3, but not through the extrinsic pathway.
In general, there is a close correlation between apoptosis and the cell cycle. If the cell cycle is blocked in a phase, apoptosis appears in the phase in which the cell cycle is blocked[1]. In the present study, an accumulation of cells in the S phase was observed immediately after the treatment of CSE (Figure 7). Our findings indicate that CSE inhibits or arrests DNA replication in HepG2 cells in the S phase and then signals for apoptosis before the cells enter the G2 phase. Cyclins and their partners, cyclin-dependent kinases (CDK), constitute the basis of these molecular mechanisms. CDK2 is activated by cyclin E and allows progression into the S phase. CDK2 is also activated by cyclin A to allow progression into the S phase[24]. The effect of CSE on the activation of CDK2 and the activity of cyclin A in human hepatoma carcinoma HepG2 cells requires further investigation. The tumor suppressor protein p53 is a crucial protein in cellular stress responses. Upon DNA damage, p53 protein expression increases, and then transactivates its downstream target genes, inducing cell cycle arrest, DNA repair, and apoptosis[25,26]. It was reported that ethylnitrosourea induces neural progenitor cell apoptosis after S-phase accumulation in a p53-dependent manner[27]. Here, we showed that in HepG2 cells possessing wild-type p53, CSE-induced S-phase arrest and apoptosis were accompanied by the upregulation of p53 mRNA and protein level in a dose-dependent manner (Figures 8C, 9C). These results indicated that the activation of the p53 pathway may be involved in CSE-induced cell cycle arrest and apoptosis in HepG2 cells.
Cell apoptosis is a programmed death process, which is induced and controlled by many complicated factors, such as blockage of the cell cycle, changes of expression of correlative apoptosis genes, and the elevation of caspase activity[1]. Many anticancer drugs perform their curative effect by inducing apoptosis of tumor cells through those pathways. In conclusion, the data reported here indicate that CSE inhibits growth and proliferation by arresting the cell cycle at the S phase, and induces apoptosis of human hepatoma HepG2 cells. This apoptosis was mediated by the activation of intrinsic caspase cascade, the downregulation of the Bcl-2 level, and the upregulation of Bax and the p53 level in HepG2 cells. The results of the present study provide supportive data for the anticancer potential of CSE.
References
- Song GB, Luo Q, Qin J, Wang L, Shi YS, Sun CX. Effects of oxymatrine on proliferation and apoptosis in human hepatoma cells. Colloid Surface B 2006;48:1-5.
- Ji XY, Tan BKH, Zhu YZ. Salvia miltiorrhiza and ischemic diseases. Acta Pharmacol Sin 2000;21:1089-94.
- Ryu SY, Lee CO, Choi SU. In vitro cytotoxicity of tanshinone from Salvia miltiorrhiza. Planta Med 1997;63:339-42.
- Liu J, Shen HM, Ong CN. Salvia miltiorrhiza inhibits cell growth and induces apoptosis in human hepatoma HepG2 cells. Cancer Lett 2000;153:85-93.
- Zhang H, Yu C, Jia JY, Leung SW, Siow YL, Man RY, et al. Contents of four active components in different commercial crude drugs and preparations of danshen (Salvia miltiorrhiza). Acta Pharmacol Sin 2002;23:1163-8.
- Gao YQ, Wang H, Jiang MC, Guo J. Effect of salvianolic acid B on proliferation of K562 Human cell line in vitro. Acta Acad Med CPAPF 2005;14:171-3.
- Goto H, Shimada Y, Akechi Y, Kohta K, Hattori M, Terasawa K. Endothelium-dependent vasodilator effect of extract prepared from the roots of Paeonia lactiflora on isolated rat aorta. Planta Med 1996;62:436-9.
- Okubo T, Nagai F, Seto T, Satoh K, Ushiyama K, Kano I. The inhibition of phenylhydroquinone-induced oxidative DNA cleavage by constituents of Moutan Cortex and Paeoniae Radix. Biol Pharm Bull 2000;23:199-203.
- Tsuboi H, Hossain K, Akhand AA, Takeda K, Du J, Rifai M, et al. Paeoniflorin induces apoptosis of lymphocytes through a redox-linked mechanism. J Cell Biochem 2004;93:162-72.
- Lee SMY, Li MLY, Tse YC, Fung KP, Lee CY, Waye MMY. Paeoniae Radix a Chinese herbal extract inhibits hepatoma cells growth by inducing apoptosis in a p53 independent pathway. Life Sci 2002;71:2267-77.
- Frankfurt OS, Krishan A. Apoptosis-based drug screening and detection of selective toxicity to cancer cells. Anti-cancer Drug 2003;14:555-61.
- Lee SJ, Ko WG, Kim JH, Sung JH, Lee SJ, Moon CK, et al. Induction of apoptosis by a novel intestinal metabolite of ginseng saponin via cytochrome c-/mediated activation of caspase-3 protease. Biochem Pharmacol 2000;60:677-85.
- Cheng YL, Change WL, Lee SC, Liu YG, Chen CJ, Lin SZ, et al. Acetone extract of Angelica sinensis inhibits proliferation of human cancer cells via inducing cell cycle arrest and apoptosis. Life Sci 2004;75:1579-94.
- Paola S, Riccardo G. Resveratrol as an anticancer nutrient: molecular basis, open questions and promises. J Nutr Biochem 2005;16:449-66.
- Morre DJ, Morre DM. Synergistic capsicum tea mixture with anticancer activity. J Pharm Pharmacol 2003;55:987-94.
- Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z, Roskams T, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004;40:667-76.
- Nishikawa T, Nakajima T, Moriguchi M, Jo M, Sekoguchi S, Ishii M, et al. A green tea polyphenol, epigalocatechin-3-gallate, induces apoptosis of human hepatocellular carcinoma, possibly through inhibition of Bcl-2 family proteins. J Hepatol 2006;44:1074-82.
- Huether A, Hopfner M, Sutter AP, Schuppan D, Scherübl H. Erlotinib induces cell cycle arrest and apoptosis in hepatocellular cancer cells and enhances chemosensitivity towards cytostatics. J Hepatol 2005;43:661-9.
- Maaser K, Sutter AP, Scherübl H. Mechanisms of mitochondrial apoptosis induced by peripheral benzodiazepine receptor ligands in human colorectal cancer cells. Biochem Bioph Res Commun 2005;332:646-52.
- Zhang JF, Liu JJ, Liu PQ, Lin DJ, Li XD, Chen GH. Oridonin inhibits cell growth by induction of apoptosis on human hepatocelluar carcinoma BEL-7402 cells. Hepatol Res 2006;35:104-10.
- Michael H, Andreas PS, Alexander H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J Hepatol 2004;41:1008-16.
- Waxman DJ, Schwartz PS. Harnessing apoptosis for improved anticancer gene therapy. Cancer Res 2003;63:8563-72.
- Ho FY, Tsang WP, Kong SK, Kwok TT. The critical role of caspases activation in hypoxia/reoxygenation induced apoptosis. Biochem Biophys Res Commun 2006;345:1131-7.
- Schang LM. Effects of pharmacological cyclin-dependent kinase inhibitors on viral transcription and replication. BBA-Proteins Proteom 2004; 1697: 197–209.
- Yu J, Zhang L. The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Commun 2005;331:851-8.
- Shi MX, Cai QF, Yao LM, Mao YB, Ming YL, Ou-Yang GL. Antiproliferation and apoptosis induced by curcumin in human ovarian cancer cells. Cell Biol Int 2006;30:221-6.
- Katayama K, Masaki U, Hirofumi Y, Takayuki N, Hiroyuki N, Kunio D. Ethylnitrosourea induces neural progenitor cell apoptosis after S-phase accumulation in a p53-dependent manner. Neurobiol Dis 2005;18:218-25.