
Inhibitory effect of ginsenoside Rb1 on calcineurin signal pathway in cardiomyocyte hypertrophy induced by prostaglandin F2α1
Introduction
Cardiac hypertrophy is recognized as an adaptive response characterized by the growth of individual cardio-myocytes in size rather than the increase in cell number. Initially beneficial, sustaining cardiac hypertrophy eventually leads to decompensation and results in dilated cardio-myopathy, arrhythmia, fibrotic disease, heart failure, and even sudden death[1]. Furthermore, some studies have indicated that hypertrophy may not be required for a successful adaptation to increased workload[2]. Clinical studies have found that several classes of drugs, including angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, calcium channel blockers[3–5], and nitric oxide (NO)[6] could have some beneficial effects in the prevention or treatment of cardiac hypertrophy. However, more studies should be done to provide more therapeutic choices for cardiac hypertrophy.
It has been reported that ginsenosides extracted from the root of the herb Panax ginseng CA. Mey have many pharmacological effects, including increasing the activity of superoxide dismutase[7] and protecting the brain and heart from ischemic and reperfusion injuries[8,9]. Notably, ginsenoside Rb1 (Rb1), a major component in ginsenosides, has been shown to elevate the release of NO in rat ventricular myocytes[10] and decrease intracellular free Ca2+ in cardiac myocytes and other tissues[11–13], which indicates that Rb1 may be a potential drug for anticardiac hypertrophy.
Prostaglandin F2α (PGF2α) has been shown to induce cardiac myocyte hypertrophy in vitro and cardiac growth in vivo and is a good candidate to mediate the growth of cardiac cells[14]. Meanwhile, Ca2+ signaling has been reported to play a critical role in the development of cardiac hypertrophy induced by various hypertrophic stimuli[15]. The increased intracellular Ca2+ binds to calmodulin and regulates several downstream effectors, such as calcineurin (CaN), which is a key mediator of cardiac hypertrophy[16,17]. CaN dephosphorylates the nuclear factor of activated T cells (NFAT), and then later translocates to the nucleus where it acts with other transcription factors (eg GATA4) for the activation of downstream target genes to induce cardiac hypertrophy[18,19]. A series of studies has shown that the neuro-protective activities and anti-aging function of Rb1 were related to decreasing intracellular Ca2+[11–13]. However, whether Rb1 has antihypertrophic effects on cardiac hypertrophy and inhibits the Ca2+–CaN signal pathway has not known as yet.
Materials and methods
Primary culture of myocytes Ventricular myocytes from 1–3-d-old rats (Animal Center of Institute of Surgery Research of the Third Military Medical University, Chongqing, China) were prepared and cultured for 48 h in Dulbecco’s modified Eagle’s medium (DMEM) containing 20% fetal bovine serum and 0.1 mmol/L bromodeoxyuridine (Sigma, St Louis, Missouri, USA) as described previously[20]. The cells were adjusted to 1.5×106–3×106 cells/mL for measuring intracellular free calcium concentration ([Ca2+]i), or to 0.5×106–1×105 cells/mL for measuring cell diameter and protein content. The medium was replaced by serum-free DMEM for a further 48 h before the treatment of drugs. 100 nmol/L PGF2α (Cayman Chemical, Ann Arbor, Michigan, USA) was used to stimulate the cardiomyocytes, Rb1 with 99% purity and final concentrations of 50, 100, and 200 µg/mL (Division of Chinese Material Medical and Natural Products, National Institute for the Control of Pharmaceutical and Biological Products, Ministry of Public Health, Beijing, China) and L-arginine with final concentration of 1 mmol/L (Alexis, Lausen, Switzerland) were used to investigate their antihypertrophic effects. NG-nitro-L-arginine-methyl ester (L-NAME; Alexis, Lausen, Switzerland) 1 mmol/L was used to investigate the relationship between the antihypertrophic effects of Rb1 and NO.
Measurement of cardiomyocytic diameters The cardio-myocytes were fixed in 4% polyformaldehyde solution and stained with HE. The diameter of single cells was measured by the BI2000 Imaging Analytic System (Chengdu Taimeng Sci-Tec, Chengdu, China). Five random fields (10−15 cells per field) from each slide were analyzed. The experiments were repeated 3 times.
Measurement of cardiomyocytic protein contents The cardiomyocytes were digested by trypsinase and counted. The cells were then washed 3 times with Hanks’ balanced salt solution (HBSS) without Ca2+ and Mg2+ (D-HBSS; in mmol/L: NaCl 137.0, KCl 5.0, Na2HPO4 0.6, KH2PO4 0.4, NaHCO3 3.0, glucose 5.6, pH 7.2) by centrifuging at 400×g for 2 min. The cardiomyocytes were homogenized with RIPA lysis buffer (Upstate, Lake Placid, New York, USA) and centrifuged at 12 000×g for 20 min at 4 °C. The protein concentration in the supernatant was determined by the Bradford assay using bovine γ-globulin as the standard, then the protein concentration per cell was calculated.
Measurement of [Ca2+]i The [Ca2+]i was measured by the method described before[21]. Briefly, the cells (1×106) were incubated in the medium with Fura 2/AM (5 µmol/L; Sigma, St Louis, USA) for 50 min at 37 °C, then washed 3 times with HBSS (D-HBSS plus 1.30 mmol/L CaCl2 and 0.5 mmol/L MgCl2) containing 0.2% bovine serum albumin by centrifuging at 500×g for 2 min. The fluorescence value from 1 mL cell suspension was measured by a Shimadzu RF-5000 spectrofluorometer (Kyoto, Japan) with dual excitation wavelengths at 340 and 380 nm and emission wavelengths at 510 nm. The [Ca2+]i was calculated by the following equation: [Ca2+]i=Kd×(F–Fmin)/(Fmax–F). Here, Kd was the dissociation constant of Fura 2/AM for Ca2+ (about 224 nmol/L at 37 °C), F was the basal fluorescence value of the cells, Fmax was the fluorescence value under the presence of excess calcium in the cells due to the lysis of the cellular membrane caused by 0.98 g/L Triton-X 100 (Sigma, St Louis, Missori, USA), Fmin was the fluorescence value under the presence of minimal calcium using 5 mmol/L ethyleneglycotetraacetic acid (EGTA) to chelate the Ca2+ in the cells after the lysis of the cellular membrane by Triton-X 100.
RT-PCR analysis of mRNA Total RNA was extracted from the cardiomyocytes by use of an RNeasy mini kit (Qiagen, Valencia, California, USA). RT-PCR was performed with an RT-PCR kit (Promega, San Jose, California, USA) according to the manufacturer’s instructions. The nucleotide sequence of the primers were as follows[22,23]: (i) atrial natriuretic peptide (ANP): sense 5'- GCC CTG AGC GAG CAG ACC GA -3', antisense 5'-CGG AAG CTG TTG CAG CCT A-3'; (ii) CaN: sense 5'-ACT GGC ATG CTC CCC AGC GGA-3', antisense 5'-GTG CCG TTA GTC TCT GAG GCG-3'; and (iii) β-actin: sense 5'-GAC TAC CTC ATG AAG ATC CTG ACC-3', antisense 5'-TGA TCT TCA TGG TGC TAG GAG CC-3'. The predicted products in size were 202, 244, and 423 bp, respectively. These primers were synthesized by Beijing Dingguo Biotech (Beijing, China). The following conditions of the RT-PCR reactions were met: (i) 1 cycle at 48 °C for 45 min, 94 °C for 2 min; (ii) 35 cycles at 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min; and (iii) 72 °C for 7 min. The products were separated by electrophoresis on 1% agarose gel containing ethidium bromide, and photographed. The integral optical density values for each band of ANP, CaN, and β-actin on the gel were assayed by the BI2000 Imaging Analysis System (Chengdu Taimeng Sci-Tec, China). β-actin was used as an internal control for the semiquantitative assay.
Western blotting The protein (30 µg) from cardiomyo-cytes separated by 10% SDS-PAGE was transferred onto polyvinylidene difluoride nylon membranes. The blots were probed with mouse anti-CaNA-α (1:200 dilution) or anti-NFAT3 (1:100 dilution) or anti-GATA4 antibodies (1:100 dilution; (Santa Cruz Biotechnology, Santa Cruz, California, USA), and then with horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G (1:2500 dilution) antibodies. Immunodetection was carried out using the BI2000 Imaging Analysis System.
Statistical analysis All of the data were expressed as mean±SD and analyzed by either ANOVA or Student’s t-test with SPSS 11.5 software (SPSS Inc, Chicago, Illinois, USA. Differences were considered statistically significant at P<0.05.
Results
Effects of Rb1 on PGF2α-induced cardiomyocyte hypertrophy Light microscopic findings of the cardiomyocytes showed that the cardiomyocytes treated with PGF2α (100 nmol/L) became swollen and enlarged with undistinguishable borders among the cells (Figure 1B). Rb1 (200 µg/mL) markedly alleviated the morphological changes induced by PGF2α (Figure 1C). The addition of L-NAME (1 mmol/L) could not antagonize the effect of Rb1 on the hypertrophic myocyte (data not shown).
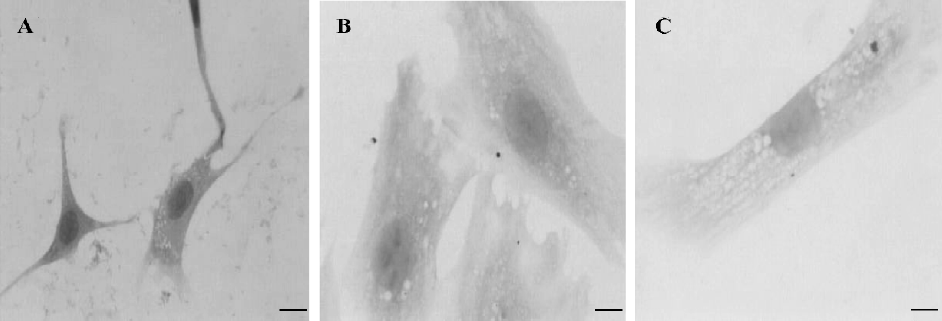
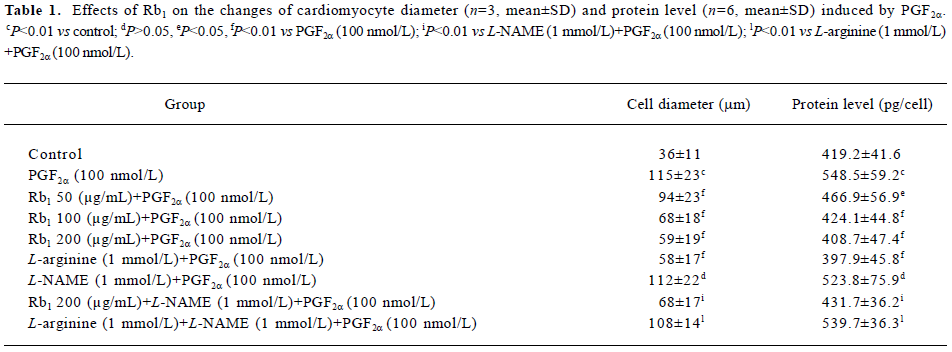
Table 1 showed that the diameters and protein contents of the cardiomyocytes treated with PGF2α significantly increased, compared with that of the control (P<0.01). The treatment of Rb1 with a variety of concentrations (50, 100, and 200 µg/mL) significantly relieved the changes induced by PGF2α in a concentration-dependent manner (P<0.05). L-arginine (1 mmol/L) also lowered these changes induced by PGF2a (P<0.01). L-NAME (1 mmol/L) abolished the effects of L-arginine, but failed to abolish the effects of Rb1 (200 µg/mL) on the cardiomyocyte diameter and protein content.
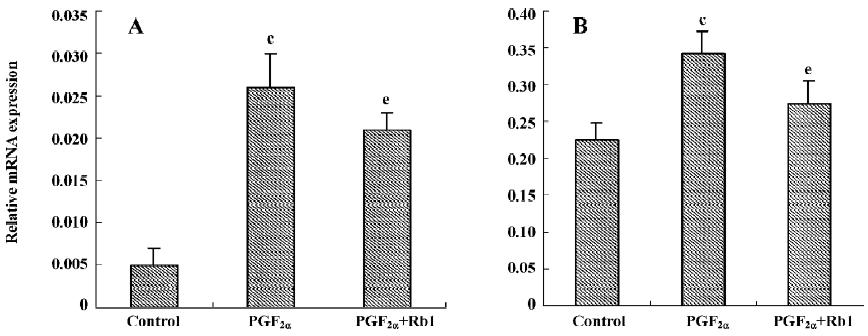
There was a low fundamental expression of ANP mRNA in the cardiac myocytes (0.005±0.002, n=3). PGF2α treatment obviously increased the ANP mRNA expression, which was significantly antagonized by Rb1 (200 µg/mL) treatment (Figure 2A).
Full table
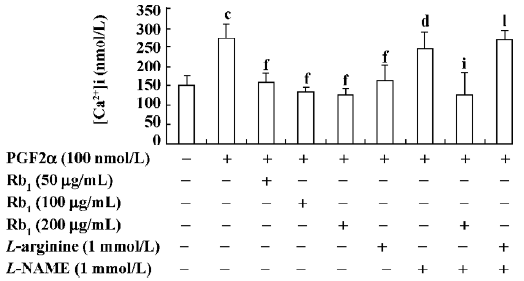
Effects of Rb1 on PGF2α-induced [Ca2+]i in cardio-myocytes The resting [Ca2+]i was 149.7±26.2 nmol/L (n=6), and it increased by 83% after the cardiomyocytes were treated with PGF2α (100 nmol/L) for 48 h (P<0.01). Treatments with either Rb1 at the concentrations of 50, 100, and 200 µg/mL or L-arginine (1 mmol/L) strongly blocked the [Ca2+]i increase caused by PGF2α. Once again, L-NAME (1 mmol/L) treatment abolished the effect of L-arginine (P<0.01), but failed to antagonize the effect of Rb1 (P>0.05; Figure 3).
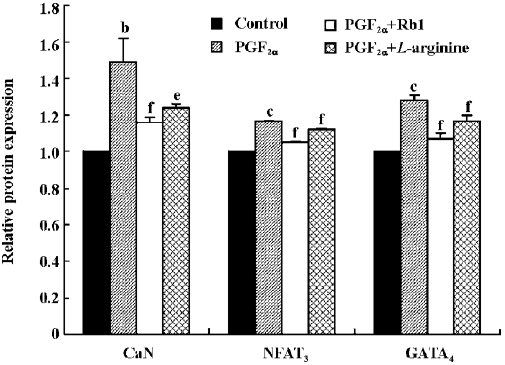
Effects of Rb1 on transcription of CaN and expressions of CaN, NFAT3, and GATA4 proteins from cardiomyocytes treated by PGF2α The relative CaN mRNA expression was 0.225±0.023 in the control and increased by 52% in the PGF2α-treated cardiomyocytes (Figure 2B; n=3, P<0.01). Similar treatments with PGF2α also significantly increased the expressions of the CaN, NFAT3, and GATA4 proteins of cardiomyo-cytes. The treatment of Rb1 (200 µg/mL) markedly decreased the mRNA expression of CaN and the protein expressions of CaN, NFAT3, and GATA4 from cardiomyocytes treated by PGF2α. L-arginine (1 mmol/L) also inhibited the protein expressions of CaN and its downstream factors (P<0.05; Figure 4, n=3).
Discussion
It has been reported that the morphological changes of cardiomyocyte hypertrophy can be induced in vitro by stimulating cultured neonatal cardiomyocytes with various growth factors and cytokines, such as angiotensin II, endothelin-1, and PGF2α, which was similar to those induced by pressure or volume load[14,24–26]. The characteristic phenotype of hypertrophy following growth factor stimulation includes an increase of cell volume and protein synthesis with an accumulation of contractile proteins, organization of the contractile proteins into sarcomeric units, as well as the re-expression of fetal cardiac genes, including ANP[27]. In this study, the findings from measuring the diameter, protein content, and ANP mRNA expression of the cardiac myocytes suggested that PGF2α could induce cardiomyocyte hypertrophy resembling that described by Lai et al[14], and that Rb1 could significantly decrease the elevated cardiomyo-cyte volume, protein content, and ANP mRNA expression caused by PGF2α.
Our results showed that PGF2α induced cardiomyocyte hypertrophy with the elevating [Ca2+]i level. Meanwhile, the antihypertrophic effects of Rb1 were accompanied simultaneously with the alleviating [Ca2+]i level. L-NAME, an NO synthase inhibitor, did not antagonize both effects of Rb1 on hypertrophy and [Ca2+]i of cardiomyocytes; on the contrary, it abolished the antihypertrophic and decreasing [Ca2+]i effects of L-arginine. These results suggested that the directly decreasing [Ca2+]i effect rather than the NO release may be responsible for the antihypertrophic effects of Rb1.
In the past several years, a number of experiments have implicated that the CaN signal transduction pathway may play an important role in the cardiomyocyte hypertrophy process[28]. We have previously reported that cardiac hypertrophy by PGF2α may be mediated by the CaN signal pathway in rats[29]. In the present paper, the fact that the transcription and expression of CaN, as well as the expression of the CaN downstream factors increased with elevating [Ca2+]i under the stimulation of PGF2α to the cardiomyocytes, which were blunted by Rb1, suggested that the interference of the CaN signaling pathway might be involved in the antihyper-trophic mechanisms of Rb1.
L-arginine, an NO donor, was observed to inhibit cardio-myocyte hypertrophy[30], which could be abolished by L-NAME. Surprisingly, L-NAME had no influence on either the antihypertrophic effect or on the inhibiting [Ca2+]i rise from Rb1. The results suggested that the antihypertrophic effect of Rb1 might be different from L-arginine, but the relationship between the Rb1 effects and NO still needs much investigation.
In conclusion, our study demonstrates that Rb1 can alleviate cardiac hypertrophy in vitro, which may be mediated in part by an inhibitive effect on elevated [Ca2+]i due to the inactivation of the CaN transduction pathway.
References
- Hunter JJ, Chien KR. Signaling pathways for cardiac hypertrophy and failure. N Engl Med 1999;341:1276-83.
- Esposito G, Rapacciuolo A, Naga Prasad SV, Takaoka H, Thomas SA, Koch WJ, et al. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevents cardiac dysfunction despite increased wall stress. Circulation 2002;105:85-92.
- Jacobs TF, Ramsey LA. Angiotensin receptor blockers. Applications beyond hypertension. Adv Nurse Pract 2005;13:27-30.
- Zhang R, Crump J, Reisin E. Regression of left ventricular hypertrophy is a key goal of hypertension management. Curr Hypertens Rep 2003;5:301-8.
- Messerli FH. Evolution of calcium antagonists: past, present, and future. Clin Cardiol 2003;26 Suppl 2:II12-6.
- Kempf T, Wollert KC. Nitric oxide and the enigma of cardiac hypertrophy. Bioassays 2004;26:608-15.
- Zhang JL, Xu WA, Yang YK, Zhang EY, Wang ZY, Zhang YZ. A primarily study on the relationship between the effect of Panax Notoginsen Rb1 Rg1 on the aging process. Acad J Kunming Med 2000;21:63-5. Chinese..
- Jiang S, Jiang ZL. Protective effect of ginsenoside Rb1 on ischemic brain injury in rat. J Apoplexy Nervous Diseases 2003;20:415-7.
- Guan L, Li W, Liu Z. Effect of ginsenoside Rb1 on cardiomyocyte apoptosis after ischemia and reperfusion in rats. J Huazhong Univ Sci Technol Med Sci 2002;22:212-5. Chinese..
- Scott GI, Colligan PB, Ren BH, Ren J. Ginsenosides Rb1 and Re decrease cardiac contraction in adult rat ventricular myocytes: role of nitric oxide. Br J Pharmacol 2001;134:1159-65.
- Jiang XY, Zhang JT, Shi CZ. Mechanism of action of ginsenoside Rb1 in decreasing intracellular Ca2+. Yao Xue Xue Bao 1996;31:321-6. Chinese..
- Liu M, Zhang JT. Effects of ginsenoside Rb1 and Rg1 on synaptosomal free calcium level, ATPase and calmodulin in rat hippocampus. Chin Med J 1995;108:544-7.
- Zhang WJ, Zhong GG, Jiang Y, Wang XM, Wang ZF. Single channel analysis on calcium channel blocked action of panaxadiol and panaxatriol saponins on cultured rat ventricular myocytes. Acta Pharmacol Sin 1994;15:173-6. Chinese..
- Lai J, Jin H, Yang R, Winer J, Li W, Yen R, et al. Prostaglandin F2 alpha induces cardiac myocyte hypertrophy in vitro and cardiac growth in vivo. Am J Physiol 1996;271:H2197-208.
- Sugden PH. Signaling pathways in cardiac myocyte hypertrophy. Ann Med 2001;33:611-22.
- Zhang W. Old and new tools to dissect calcineurin’s role in pressure-overload cardiac hypertrophy. Cardiovasc Res 2002;53:294-303.
- De Windt LJ, Lim HW, Haq S, Force T, Molkentin JD. Calcineurin promotes protein kinase C and c-jun NH2-terminal kinase activation in the heart, cross-talk between cardiac hypertrophic signaling pathways. J Biol Chem 2000;275:13571-9.
- Hogan PG, Chen L, Nardone J, Rao H. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 2003;17:2205-32.
- Vega RB, Bassel-Duby R, Olson EN. Control of cardiac growth and function by calcineurin signaling. J Biol Chem 2003;278:36981-4.
- Xu SY, editor. Methodologies of pharmacological experiment. Beijing: The People’s Medical Publishing House; 2002.
- Lei KJ, Huang XN, Wu Q, Xie XL, Yu LM, Lu YF. The effects of breviscapine on platelet aggregation and intra-platelet free calcium. Acta Acad Med Zunyi 2004;27:11-3. Chinese..
- Taigen T, De Windt LJ, Molkentin JD. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci USA 2000;97:1196-201.
- Abassi Z, Brodsky S, Gealekman O, Rubinstein I, Hoffman A, Winaver J. Intrarenal expression and distribution of cyclooxy-genase isoforms in rats with experimental heart failure. Am J Physiol Renal Physiol 2001;280:F43-53.
- Ng DC, Long CS, Bogoyevitch MA. A role for the extracellular signal regulated kinase and p38 mitogen-activated protein kinases in interleukin-1 beta-stimulated delayed signal transducer and activator of transcription 3 activation, atrial natriuretic factor expression, and cardiac myocyte morphology. J Biol Chem 2001;276:29490-8.
- Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol 2003;35:851-9.
- Fujino H, Srinivasan D, Pierce KL, Regan JW. Differential regulation of prostaglandin F2α receptor isoforms by protein kinase C. Mol Pharmacol 2000;57:353-8.
- Chien KR, Grace AA, Hunter JJ. Molecular and cellular biology of cardiac hypertrophy and failure. In: Chien KR, editor. Molecular basis of cardiovascular disease. Beijing: People’s Medical Publishing House; 2001. p 211–50.
- Molkentin JD. Calcineurin and beyond: cardiac hypertrophic signaling. Circ Res 2000;87:731-8.
- Jiang QS, Huang XN, Yang GZ, Dai ZK, Zhou QX, Shi JS, et al. Signal transduction pathway of cardiac hypertrophy induced by prostaglandin F2α may be mediated by calcineurin in rats. Acta Physiol Sin 2005;57:742-8.
- Fiedler B, Wollert KC. Interference of antihypertrophic molecules and signaling pathways with the Ca2+-calcineurin-NFAT cascade in cardiac myocytes. Cardiovasc Res 2004;63:450-7.