
Prevention of lipopolysaccharide-induced injury by 3,5-dicaffeoylquinic acid in endothelial cells
Introduction
The vascular endothelium serves as the key barrier between the intravascular compartment and extravascular tissues, and plays a critical role in a large number of physiological and pathological processes[1]. The progressive dysfunction of the endothelium is regarded as a common denominator of impaired organ function after a variety of pathological tissue alterations such as ischemia, shock, or sepsis[2].
Lipopolysaccharide (LPS) is a critical glycolipid component of the outer wall of Gram-negative bacteria. LPS can induce apoptosis in bovine and ovine endothelial cells in vitro. When the expression of new genes is inhibited using cycloheximide (CHX), human endothelial cells become responsive to the apoptotic signals induced by LPS[6]. The induction of reactive oxygen species (ROS) by LPS correlates with the onset of apoptosis, and agents that inhibit the formation of ROS are known to protect against LPS-induced endothelial cell apoptosis[1,3].
The phenylethanoid 3,5-dicaffeoylquinic acid (3,5-diCQA, a white power, Mw 516.44, purified by CC and HPLC) is present in a variety of plants, including green coffee beans (Lactuca indica)[4,5], and is an important component of the Chinese herb Erigeron breviscapus. Apart from functioning as a free radical scavenger, 3,5-diCQA inhibits tyrosinase activity and has a neuroprotective capacity[5,14]. Anti-oxidant or anti-apoptotic activity has not previously been described for 3,5-diCQA in endothelial cells. In the present study, we investigated the effect of 3,5-diCQA on lipid peroxides in microsomes, LPS-stimulated ROS generation, and LPS-induced injury in human dermal microvascular endothelial cells (HMEC-1) (Figure 1).
Materials and methods
Reagents 3,5-diCQA was a gift from the Kunming Institute of Botany (Chinese Academy of Sciences, Kunming Shanghai, China). The purity of the compounds was greater than 95%, as verified by HPLC. LPS (Escherichia coli 0111:B4), CHX, BSA, 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyl-tetrazolium bromide (MTT) and MCDB-131 medium were purchased from Sigma (St Louis, MO, USA). 2,7-dichloro-dihydrofluorescein diacetate (DCFH-DA) was obtained from Beyotime Institute of Biotechnology (Haimen, China). The lactate dehydrogenase (LDH) and malondialdehyde (MDA) assay kits were purchased from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The CaspACE assay system colorimetric kit was from Promega (USA). All other reagents or drugs were of analytical grade.
Preparation of microsomes and assay of lipid peroxides The microsomes were prepared from rat livers. The liver was cut into small pieces and homogenized in cold buffer A [(0.l mol/L sucrose, 0.05 mol/L KCl, 0.04 mol/L KH2PO4, and 0.03 mol/L EDTA (pH 7.2)]. After centrifugation (1000×g, 20 min), the supernatant was transferred to another tube and recentrifuged at 105 000×g for 60 min. The sediment was washed and resuspended in buffer A. All operations were carried out at 4 °C[9]. Lipid peroxidation of microsomes was measured as described previously[17] with the following modifications: the membranes, at a concentration of 1.5 mg protein, were incubated at 37 °C with 0.05 mol/L phosphate (pH 7.4) and 0.4 mmol/L ascorbic acid. The final volume was 1.5 mL. Phosphate buffer was contaminated with sufficient iron to provide the necessary ferric iron for lipid peroxidation. Control membrane preparations were performed in parallel in the absence of ascorbate. Lipid peroxidation was initiated by incubating the membrane preparations in a water bath at 37 °C for 30 min. Lipid peroxidation was assessed by detection of thiobarbituric acid-reactive MDA, an end product of the peroxidation of polyunsaturated fatty acids and related esters. The level of MDA was determined using the MDA assay kit. The results were normalized to the level of MDA equivalents/mg of protein.
Cell culture and drug treatment HMEC-1 were maintained in a 5% CO2 atmosphere at 37 °C in MCDB-131 medium supplemented with 10% FBS and 10 µg/L human recombinant epidermal growth factor. The cells treated with CHX (50 mg/L) were used as controls unless otherwise specified. Cell injury was induced by incubation with LPS (100 µg/L) and CHX (50 mg/L). 3,5-diCQA was added to the cells 1 h prior the addition of LPS and CHX. All experiments were carried out on confluent cultures.
Assay of intracellular ROS The measurement of intracellular ROS was based on the ROS-mediated conversion of non-fluorescent DCFH-DA into DCFH. DCFH is membrane impermeable and rapidly oxidized to the highly fluorescent 2,7-dichlorofluorescein in the presence of intracellular ROS[23]. The intensity of fluorescence reflects the level of oxidative stress. Intracellular ROS was measured after incubation in LPS and/or CHX for 12 h by replacing the medium with 50 mmol/L phosphate buffer (pH 7.4) containing 10 µmol/L DCFH-DA, and incubating in the dark for 1 h at 37 °C. The level of DCFH fluorescence was measured at an emission wavelength of 530 nm and an excitation wavelength of 485 nm by flow cytometry[19]. The results were expressed as the percentage of control fluorescence intensity.
Assay of cell survival and cell damage To quantitate the proportion of viable cells, HMEC-1 were seeded onto 96-well plates at a density of 40 000 cells/well. The cells were incubated in LPS and/or CHX for 16 h. The viable cell number was estimated using the MTT assay. Briefly, the medium was removed and replaced with Krebs (120 mmol/L NaCl, 5.6 mmol/L KCl, 1.2 mmol/L MgSO4, 1.2 mmol/L NaH2PO4, 25 mmol/L NaHCO3, 10 mmol/L glucose, and 2.5 mmol/L CaCl2) containing 1% BSA and 1 g/L MTT, and then incubated for 5 h. The medium was aspirated and the formazan product was solubilized with DMSO. Absorbance at 630 nm (back-ground absorbance) was subtracted from absorbance at 570 nm for each well. Viability was expressed as a proportion of the viable cell number incubated without LPS[6,15]. Both released and total LDH concentrations were determined. The release of LDH from cells is a pathological manifestation of increased plasma membrane permeability. The total LDH was determined after incubation in the presence and absence of LPS for 16 h by releasing the compound from the cells with 1% Triton-X 100 and incubating for 30 min at 37 °C. The level of LDH was determined in samples of culture medium (30 µL) with or without Triton-X 100 using the LDH detection kit. The activity of LDH was corrected for the volume, and released LDH activity was expressed as a percentage of total cellular LDH[8].
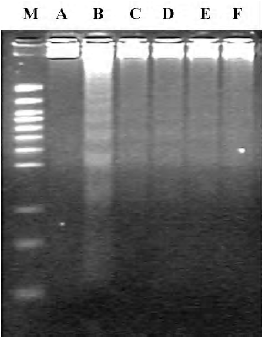
DNA fragmentation assay After incubation in the presence or absence of LPS for 16 h, the cells (5×105) were harvested by centrifugation and washed twice with phosphate buffered solution (PBS). The cells were then lysed by incubating in lysis buffer [20 mmol/L EDTA, 100 mmol/L Tris (pH 8.0), and 0.8% (w/v) SDS] with RNaseA/T1 (RNase A 500 U/mL, RNase T1 20 kU/mL (Amesco) for 2 h at 37 °C. The lysed cells were then incubated in the presence of proteinase K (20 mg/mL) for 4 h at 55 °C. DNA samples were loaded onto 2% horizontal agarose gels containing ethidium bromide. The gels were run at 35 V for 6 h, and the DNA fragments were visualized using UV illumination[16].
PI apoptosis assay For the flow cytometric analysis of fragmented DNA, 5×105 cells/well on the 6-well plates were harvested after incubation in the presence or absence of LPS for 12 h, incubated with 1 mL of 75% cold ethanol for 2 h at -20 °C, and then washed with PBS. The cell pellets were incubated with 50 mg/L PI and 100 mg/L RNase A at 37 °C for 30 min. Sub G1 cell counts were determined on a FACSort flow cytometer using the Cell Quest analysis program[7].
Caspase-3 activity assay Caspase-3 activity was evaluated using the CaspACE assay system according to the protocol provided by the manufacturer. The cells plated in the flasks were treated with LPS and/or CHX for 8 h, were then harvested, washed, and lysed by 4 cycles of freezing and thawing, followed by incubation on ice for 15 min. The samples were then centrifuged at 15 000×g for 20 min at 4 °C. The cell lysate supernatant was aliquoted into the wells of a 96-well plate, and 32 µL buffer, 2 µL DMSO, 10 µL DTT (100 mmol/L), and 2 µL of DEVD-pNA (10 mmol/L) were added to a final volume of 100 µL. The samples were incubated for 4 h at 37 °C. The absorbance at 405 nm was measured and enzyme activity was calculated as pmol pNA liberated per hour/µg protein according to the formula provided by the manufacturer.
Statistical analysis All experiments were repeated at least 3 times. All data were expressed as mean±SD. Statistical analysis was performed by the Student’s t-test using the software SPSS 11.0 for Windows (SPSS, Chicago, USA). P<0.05 was considered statistically significant.
Results
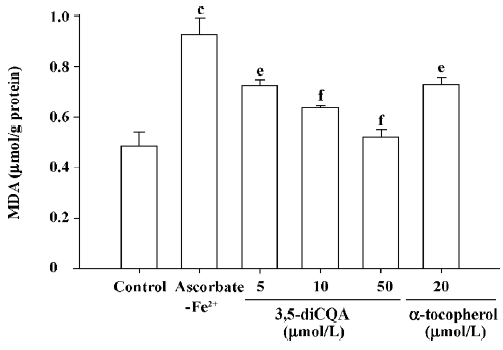
Effect of 3,5-diCQA on microsome lipid peroxidation The anti-oxidative activity of 3,5-diCQA was assessed by measuring its effect on microsome lipid peroxidation. The exposure of microsomes to ascorbate-Fe2+ led to an increase in the level of MDA from the control level of 0.49 µmol/g protein to 0.93 µmol/g protein (P<0.01) indicating lipoper-oxidation. A concentration-dependent decrease in ascorbate-Fe2+-induced MDA formation to 0.72, 0.64, and 0.52 µmol/g protein was observed for 3,5-diCQA at 5, 10, and 50 µmol/L, respectively (Figure 2). Treatment with 20 µmol/L α-tocopherol, a well known anti-oxidant, gave an MDA value of 0.73 µmol/g protein (P<0.01).
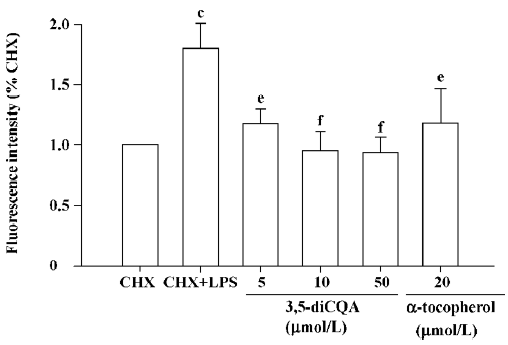
Inhibitory effect of 3,5-diCQA on ROS formation According to the result in the model of the microsome above, we further determined intracellular ROS levels to assess the anti-oxidative activity of 3,5-diCQA in LPS-stimulated HMEC-1. Cells treated with CHX and LPS for 12 h gave an increase in intensity of fluorescence indicative of ROS formation, 1.8-fold the intensity observed for the control cells treated with CHX alone. Pretreatment with 3,5-diCQA (5, 10, or 50 µmol/L) or α-tocopherol (20 µmol/L) significantly decreased the intensity of fluorescence (Figure 3).
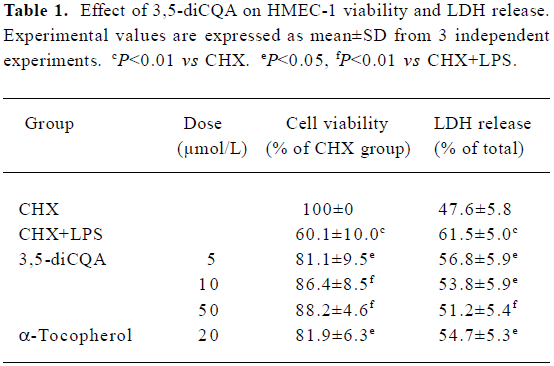
Effect of 3,5-diCQA on cell viability and damage figure Consistent with previously reported data, and supported by the predictive assay presented in the present report, LPS did not induce injury in HMEC-1 alone. Injury was induced when HMEC-1 were treated with LPS together with CHX. The effect of 3,5-diCQA on LPS-induced apoptosis in HMEC-1 in the presence of CHX was examined. After 16 h incubation with both LPS and CHX, cell viability was reduced to 60% of the level of the control group treated with CHX alone. Pretreatment with 3,5-diCQA (5, 10, or 50 µmol/L) decreased the cell death rate to 19%, 14%, and 12%, respec-tively (Table 1). The release of LDH after LPS treatment for 16 h was significantly higher than the cells treated with CHX alone. Treatment with 3,5-diCQA at 5, 10, or 50 µmol/L decreased LDH release in a concentration-dependent manner. α-Tocopherol (20 µmol/L) also showed significant suppression of LPS-induced cell death and LPS-induced LDH release (Table 1).
Full table
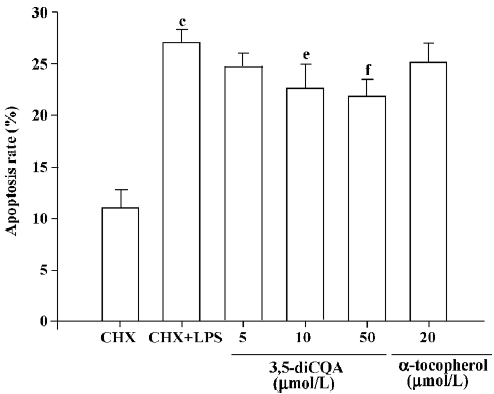
Effect of 3,5-diCQA on cell apoptosis Apoptosis induced by LPS in the presence of CHX was confirmed by the DNA fragmentation assay. CHX alone did not induce fragmentation of chromosomal DNA. Treatment with 3,5-diCQA or α-tocopherol suppressed LPS-induced DNA laddering (Figure 4). Flow cytometric analysis was performed after PI staining. DNA fragmentation was observed in 27% of cells with hypodiploid DNA in LPS-induced apoptosis. The percentage of cells with DNA fragmentation decreased significantly on treatment with 10 or 50 µmol/L 3,5-diCQA, while 5 µmol/L 3,5-diCQA and 20 µmol/L α-tocopherol had no effect (Figure 5).
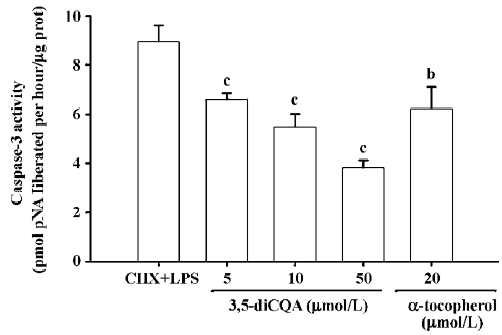
Effect of 3,5-diCQA on caspase-3 activity Caspase-3 activity in HMEC-1 was 8.92 pmol pNA liberated/hour in LPS-induced apoptosis. In the cells treated with CHX alone, the caspase-3 activity was similar to the control group (data not shown). Caspase-3 activity decreased in a dose-dependent manner on treatment with 3,5-diCQA. Caspase-3 activity induced by LPS was suppressed by 20 µmol/L α-tocopherol (Figure 6).
Discussion
In the present study, 3,5-diCQA effectively suppressed the generation of oxide free radicals in the microsome model, reduced the production of intracellular ROS induced by LPS in HMEC-1, protected HMEC-1 from LPS-induced cell injury, and inhibited the activity of caspase-3.
LPS is a bacterial endotoxin that functions as a pro-inflammatory mediator inducing significant levels of endogenous TNF-α and mediating endothelial cell damage[11]. LPS induces apoptosis in bovine and ovine endothelial cells in vitro and elicits apoptosis in human endothelial cells in the absence of new gene expression. The induction of apoptotic cell death by LPS is confirmed by several criteria, including morphological changes, DNA laddering, TdT-mediated dUTP nick end labeling, the activation of caspase, and poly (ADP-ribose) polymerase cleavage[1].
In the present study, LPS-induced cytotoxicity of HMEC-1 was suppressed by treatment of the cells with 3,5-diCQA or α-tocopherol. Cell injury, determined by the release of LDH, was also suppressed by treatment with 3,5-diCQA. The DNA fragmentation associated with LPS-induced cell death was partially inhibited by treatment with 3,5-diCQA.
Oxidants or pro-oxidants are significant regulators of apoptosis and can induce the apoptotic process[24,25]. The significance of oxidative stress in apoptosis is strongly supported by the capacity of various cellular anti-oxidants to block apoptosis induced by a diverse number of agents[25,26]. Oxidative stress is a common factor in apoptosis and is induced by stimuli including LPS and TNF-α exposure[3,19,27–29]. Several phenylethanoids also possess free radical scavenging properties and protect cells against oxidative stress-induced injury[30–32]. In the present study, we investigated the anti-oxidant capacity of the phenylethanoid 3,5-diCQA and found that both lipid peroxidation of microsomes and intracellular ROS accumulation induced by LPS were significantly suppressed. These results suggest that 3,5-diCQA may protect endothelial cells against apoptosis by directly scavenging intracellular ROS.
The signaling pathways induced by apoptosis converge to the caspase pathway to execute the final phase of the apoptotic process[12]. The caspase family of proteases consists of at least 14 members in mammals that are constitutively expressed in almost all cell types as inactive pro-enzymes (zymogens) that become processed and activated in response to a variety of pro-apoptotic stimuli[13]. Caspase-3 is a downstream member of the caspase cascade and acts as a central effector in the execution phase, the proteolytic events of which can lead to apoptosis and contribute to DNA fragmentation, nuclear morphological changes, and eventual cell death. The effect of LPS-induced caspase activation on endothelial cells is dramatic, resulting in the cleavage of nuclear proteins and structural proteins that mediate cell-cell and cell-substrate adhesion[18,21]. In vivo studies support a role for caspases in mediating LPS-induced EC apoptosis[20,22]. Thus, substances that inhibit the activity of caspase-3 may protect cells from apoptosis. The anti-apoptotic capacity of 3,5-diCQA was identified in the present study by the marked suppression of caspase-3 activity in LPS-treated cells.
In summary, 3,5-diCQA displays anti-oxidative and anti-apoptotic activity in HMEC-1 due to the scavenging of intracellular ROS and the reduction of caspase-3 activity. The anti-oxidative and anti-apoptotic capacity of 3,5-diCQA provides clinical potential for the treatment of diseases involving the progressive dysfunction of the endothelium, including ischemia, shock, or sepsis.
References
- Bannerman DD, Goldblum SE. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am J Physiol Lung Cell Mol Physiol 2003;284:L899-914.
- Schildberg FA, Schulz S, Dombrowski F, Minor T. Cyclic AMP alleviates endoplasmic stress and programmed cell death induced by lipopolysaccharides in human endothelial cells. Cell Tissue Res 2005;320:91-8.
- Kim KY, Shin HK, Choi JM, Hong KW. Inhibition of lipopolysaccharide-induced apoptosis by cilostazol in human umbilical vein endothelial cells. J Pharmacol Exp Ther 2002;300:709-15.
- Wang SY, Chang HN, Lin KT, Lo CP, Yang NS, Shyur LF. Antioxidant properties and phytochemical characteristics of extracts from Lactuca indica. J Agric Food Chem 2003;51:1506-12.
- Iwai K, Kishimoto N, Kakino Y, Mochida K, Fujita T. In vitro antioxidative effects and tyrosinase inhibitory activities of seven hydroxycinnamoyl derivatives in green coffee beans. J Agric Food Chem 2004;52:4893-8.
- Hu X, Yee E, Harlan JM, Wong F, Karsan A. Lipopolysaccharide induces the antiapoptotic molecules, A1 and A20, in microvascular endothelial cells. Blood 1998;92:2759-66.
- Lin S, Fujii M, Hou DX. Rhein induces apoptosis in HL-60 cells via reactive oxygen species-independent mitochondrial death pathway. Arch Biochem Biophys 2003;418:99-107.
- He Q, Riley RT, Sharma RP. Pharmacological antagonism of fumonisin B1 cytotoxicity in porcine renal epithelial cells (LLC-PK1): a model for reducing fumonisin-induced nephrotoxicity in vivo. Pharmacol Toxicol 2002;90:268-77.
- Nagata Y, Yonemoto M, Iwasawa Y, Shimizu-Nagumo A, Hattori H, Sawazaki Y, et al. N-[2-[N'-pentyl-(6,6-dimethyl-2,4-hepta-diynyl)amino]ethyl]-(2-methyl-1-naphthylthio)acetamide (FY-087). A new acyl coenzyme a: cholesterol acyltransferase (ACAT) inhibitor of diet-induced atherosclerosis formation in mice. Biochem Pharmacol 1995;49:643-51.
- Gutierrez VR, de la Puerta R, Catala A. The effect of tyrosol, hydroxytyrosol and oleuropein on the non-enzymatic lipid pero-xidation of rat liver microsomes. Mol Cell Biochem 2001;217:35-41.
- Egido J, Gomez-Chiarri M, Ortý’z A, Bustos C, Alonso J, Go`mez-Guerrero C, et al. Role of tumor necrosis factor-alpha in the pathogenesis of glomerular diseases. Kidney Int 1999;43:S59-64.
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science 1998;281:1312-6.
- Yan JN, Liu CX, Xu H. Caspases promoted DADAG-induced apoptosis in human leukemia HL-60 cells. Acta Pharmacol Sin 2002;23:461-6.
- Kim SS, Park RY, Jeon HJ, Kwon YS, Chun W. Neuroprotective effects of 3,5-dicaffeoylquinic acid on hydrogen peroxide-induced cell death in SH-SY5Y cells. Phytother Res 2005;19:243-5.
- Luo WB, Dong L, Wang YP. Effect of magnesium lithospermate B on calcium and nitric oxide in endothelial cells upon hypoxia/reoxygenation. Acta Pharmacol Sin 2002;23:930-6.
- Spector D, Goldman RR, Leinwand LA. Cells: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1998.
- Leaden PJ, Catalá A. Protective effect of melatonin on ascorbate-Fe2+ lipid peroxidation of polyunsaturated fatty acids in rat liver, kidney and brain microsomes: a chemiluminescence study. J Pineal Res 2005;39:164-9.
- Bannerman DD, Sathyamoorthy M, Goldblum SE. Bacterial lipopolysaccharide disrupts endothelial monolayer integrity and survival signaling events through caspase cleavage of adherens junction proteins. J Biol Chem 1998; 273: 35 371–80.
- Chan EL, Murphy JT. Reactive oxygen species mediate endotoxin-induced human dermal endothelial NF-kappaB activation. J Surg Res 2003;111:120-6.
- Deaciuc IV, Fortunato F, D’Souza NB, Hill DB, Schmidt J, Lee EY, et al. Modulation of caspase-3 activity and Fas ligand mRNA expression in rat liver cells in vivo by alcohol and lipopolysac-charide. Alcohol Clin Exp Res 1999;23:349-56.
- Bannerman DD, Tupper JC, Ricketts WA, Bennett CF, Winn RK, Harlan JM. A constitutive cytoprotective pathway protects endothelial cells from lipopolysaccharide-induced apoptosis. J Biol Chem 2001; 276: 14 924–32.
- Kawasaki M, Kuwano K, Hagimoto N, Matsuba T, Kunitake R, Tanaka T, et al. Protection from lethal apoptosis in lipopolysaccharide-induced acute lung injury in mice by a caspase inhibitor. Am J Pathol 2000;157:597-603.
- Deng M, Zhao JY, Ju XD, Tu PF, Jiang Y, Li ZB. Protective effect of tubuloside B on TNFalpha-induced apoptosis in neuronal cells. Acta Pharmacol Sin 2004;25:1276-84.
- Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol 2004;122:339-52.
- England K, Cotter TG. Direct oxidative modifications of signalling proteins in mammalian cells and their effects on apoptosis. Redox Rep 2005;10:237-45.
- Szeto HH. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J 2006;8:E277-83.
- Brigham KL, Meyrick B, Berry LC Jr, Repine JE. Antioxidants protect cultured bovine lung endothelial cells from injury by endotoxin. J Appl Physiol 1987;63:840-50.
- Liu ZG. Molecular mechanism of TNF signaling and beyond. Cell Res 2005;15:24-7.
- Feng L, Xia Y, Garcia GE, Hwang D, Wilson CB. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor-alpha, and lipopo-lysaccharide. J Clin Invest 1995;95:1669-75.
- Xiong Q, Kadota S, Tani T, Namba T. Antioxidative effects of phenylethanoids from Cistanche deserticola. Biol Pharm Bull 1996;19:1580-5.
- Xiong Q, Tezuka Y, Kaneko T, Li H, Tran LQ, Hase K, et al. Inhibition of nitric oxide by phenylethanoids in activated macro-phages. Eur J Pharmacol 2000;400:137-44.
- Sheng GQ, Zhang JR, Pu XP, Ma J, Li CL. Protective effect of verbascoside on 1-methyl-4-phenylpyridinium ion-induced neurotoxicity in PC12 cells. Eur J Pharmacol 2002;451:119-24.