
Propofol attenuates oxidative stress-induced PC12 cell injury via p38 MAP kinase dependent pathway1
Introduction
Propofol (2,6-diisopropylphenol), the active ingredient of the common general anesthetic Diprivan, has shown direct antioxidant activity conferred by the phenolic hydroxyl group in its structure. This moiety scavenges free radicals and inhibits lipid peroxidation[1]. Since oxidative stress plays an important role in the brain ischemic injury[2], the antioxidant property of propofol suggests that it may serve as a good agent to combat ischemia. Certain hypotheses have been proposed, such as a reduction in cerebral metabolism[3], potentiation of γ-aminobutyric acid-mediated inhibition[4], and restoration of the glutamate uptake impaired during injury[5,6]. Our previous study showed that propofol protected cerebral cortical and hippocampal slices against hydrogen peroxide injury at low and mid concentrations[7], but its intracellular mechanism is still worth further investigation.
Reactive oxygen species (ROS), including hydrogen peroxide (H2O2), superoxide radical, hydroxyl radical, and peroxynitrite increased, and probably would be involved in the pathogenesis of cerebral ischemic and reperfusion injury[8,9]. ROS-induced cellular events have been implicated, at least in part, in the activation of mitogen-activated protein (MAP) kinases[9,10]. Three subfamilies of MAP kinases sensitive to ROS have been identified: extracellular-signal regulated kinase 1 and 2 (ERK1/2), c-Jun N-terminal kinase (JNK), and the p38 kinase. The kinase of each subfamily modulated specific cell functions via certain signaling pathways. However, the hypothesis that ERK1/2 may be a protective signal and JNK-p38 a pro-apoptotic signal would not always hold true, and their effect may depend on the nature of the cell type, death stimulus, duration of its activation, and probably above all, the activity of other signaling pathways[11,12]. Therefore, the mechanism of these signal transduction pathways involved in ROS-mediated cell damage remains to be elucidated.
The concentration of H2O2 in healthy individuals is normally quite low. The elevation of H2O2 concentration upon cerebral ischemic and reperfusion injury could act as a significant signal[13]. H2O2 has been used frequently as an oxidative stimulus to identify redox-sensitive processes. Rat pheochromocytoma cell line PC12 is useful for studying the intracellular signaling mechanisms and are regarded as a model for catecholamine-containing neurons[14]. In the present study, we used PC12 cells injured by H2O2 as an oxidative stress model in vitro to examine the neuroprotective effect of propofol and its intracellular mechanism, especially focusing on the roles of MAP kinases in the events.
Materials and methods
Chemicals Propofol and H2O2 were purchased from Sigma Chemicals (St Louis, MO, USA). Anti-pan- and phospho-ERK1/2 (Thr202/Tyr204), p38 (Thr180/Tyr182), and JNK (Thr183/Tyr185) antibodies and SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole] were from Cell Signaling Technology (Beverly, MA, USA). All other chemicals were commercial products of reagent grade except where indicated.
Cell cultures and determination of cell viability with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide- thiazolyl blue (MTT)reduction The PC12 cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% (v/v) donor horse serum, 5% (v/v) newborn calf serum, and antibiotics (100 units/mL penicillin and 100 mg/mL streptomycin) in flasks precoated with collagen. The cultures were maintained in a humidified atmosphere containing 5% CO2 at 37 °C. The cells were grown to 70%–80% confluence in 60 and 100 mm dishes and the growth was arrested by incubation in serum-free DMEM for 24 h prior to use. All experiments were performed with growth-arrested cells to minimize basal MAP kinase activity. The neuropro-tective effect of propofol on PC12 cell death induced by H2O2 was investigated by using [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromidethiazolyl blue] (MTT) assay (Sigma, USA). The cells were treated with H2O2 (200 µmol/L) for 24 h with or without propofol, and the cellular MTT reduction was measured as described previously[15].
Determination of apoptotic cell death with Hoechst 33258 staining and a fluorescence-activated cell sorter To further evaluate the effect of propofol on the H2O2-induced apoptosis of PC12 cells, we examined apoptotic nuclei staining and DNA fragmentation in H2O2-administered PC12 cells. After treatment with H2O2 (200 µmol/L) for 24 h with or without propofol, the cells were stained with Hoechst 33258 (Molecular Probes, Eugene, OR, USA) and visualized by fluorescence microscopy to evaluate morphological changes of the nuclei as a measure of apoptosis. In addition, separate groups of cells were also harvested and stained with Annexin V and PI (propidium iodide) (BD Biosciences, San Jose, CA, USA) to evaluate the percentage of apoptotic cells using flow cytometry.
Measurement of caspase-3 activity The PC12 cells in serum-free DMEM (60 mm collagen-coated dishes) were pre-incubated with different concentrations of propofol for 30 min, then incubated in the presence of H2O2 (200 µmol/L) for 4 h. The assay was performed according to the manufacturer’s protocol. In brief, the lysates were centrifuged at 25 000×g for 3 min at 4 °C to precipitate cell debris. 50 µL of 2×reaction buffer/Dithiothreitol mix was added to the supernatant, and finally, 5 µL of 1 mmol/L caspase-3 substrate Asp-Glu-Val-Asp (DEVD) conjugated to 7-amino-4- trifluoromethylcoumarin (AFC) was added to each tube and incubated for 1 h at 37 °C. After transferring the samples and standard solution to a 96-well microplate, we measured fluorescent activity using a fluorimeter with a 400 nm excitation filter and a 505 nm emission filter.
Measurement of the p38 MAP kinase, JNK, and ERK1/2 phosphorylations in PC12 cells MAP kinase phosphorylation induced by H2O2 in PC12 cells were performed by Western blotting. ERK1/2, JNK, and p38 activations in the cells were determined by using phospho-ERK1/2, phospho-p38 and phospho-JNK antibodies. Total ERK1/2, p38 and JNK protein expressions were measured in the presence of pan-ERK1/2, p38, and JNK antibodies. The PC12 cells in serum-free DMEM were treated with or without propofol and then incubated for 30 min. After incubation with H2O2 for 10 min, the cells were collected and lysed for the immunoblot analysis. Immunoreactive bands were visualized using enhanced chemiluminescence (ECL, Pierce, Rockford, IL, USA) and were quantified by densitometry in the linear range of film exposure using a UMAX Astra 2200 scanner and NIH Image Version 1.60 software (NIH Division of Computer Research and Technology).
Statistical analysis Data were expressed as mean±SD. Differences were analyzed for significance by Student’s t-test. The results were considered significant at P<0.05.
Results
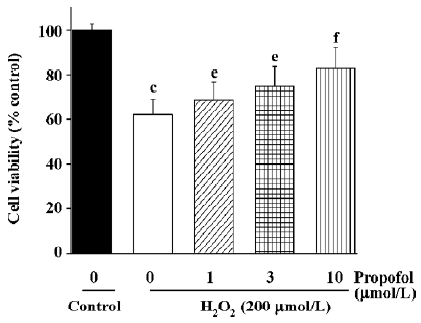
Inhibition of propofol on H2O2-induced PC12 cell death First, we examined the effect of propofol on H2O2-induced PC12 cell death by measuring MTT reduction. As shown in Figure 1, the application of H2O2 (200 µmol/L) to PC12 cell for 24 h resulted in about 35% death. Pretreatment of cells with propofol resulted in inhibition of H2O2-induced cell death in a concentration-dependent manner. No effect of propofol on cell viability was observed (data not shown).
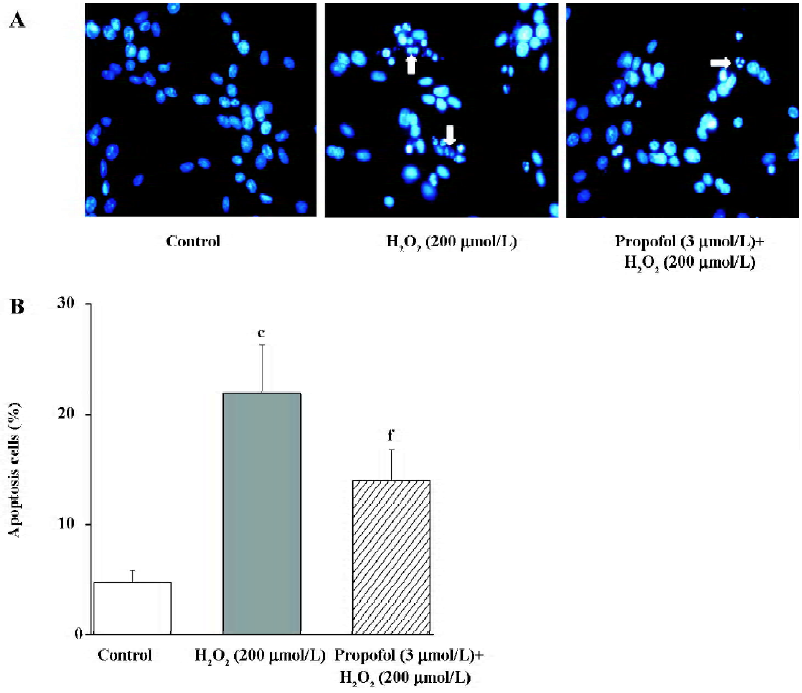
Effect of propofol on H2O2-induced PC12 cell apoptosis To evaluate the effect of propofol on the apoptosis of PC12 cells, we examined the staining of apoptotic nuclei and DNA fragmentation in H2O2-administered PC12 cells with or without propofol pretreatment. As shown in Figure 2A, staining with Hoechst 33258 revealed that PC12 cells showed apoptotic nuclei after treatment with H2O2 (200 µmol/L) for 24 h. Pretreatment with propofol decreases the rate of apoptotic nuclei. As shown in Figure 2B, in the determination of apoptotic cell death by a fluorescence-activated cell sorter (FACS), 22% of cells showed apoptosis. Pretreatment of PC12 cells with propofol (3 µmol/L) resulted in the inhibition of H2O2-induced apoptosis to 14% of total cells. The observation was consistent with that of the MTT test and the results suggested that propofol attenuates H2O2-induced PC12 cell death, including apoptosis.
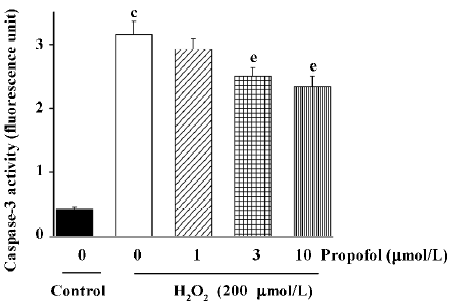
Effects of propofol on H2O2-induced caspase-3 activation in PC12 cells Since caspase-3 has been shown to be an important regulator of apoptotic cell death, we next examined the effect of H2O2 (200 µmol/L) on caspase-3 activity in PC12 cells. As shown in Figure 3, the incubation of PC12 cells with H2O2 (200 µmol/L) for 4 h significantly increased caspases-3 activity. Pre-incubation with different concentrations of propofol for 30 min significantly inhibited H2O2-induced caspase-3 activation in a concentration-dependent manner (Figure 3).
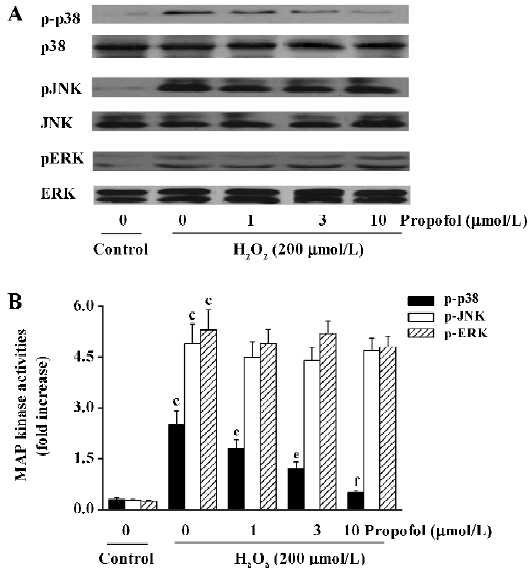
Effect of propofol on H2O2-induced ERK1/2, JNK, and p38 activation in PC12 cells To clarify the effect of propofol on H2O2-induced MAP kinase phosphorylation, the PC12 cells were incubated in the presence of H2O2 (200 µmol/L) for 10 min following pretreatment with indicated concentra-tions of propofol for 30 min. We found that H2O2-induced phosphorylation of p38 instead of JNK and ERK1/2 was inhibited by the treatment of propofol (Figure 4), which indicated that the H2O2-induced p38 MAP kinase phosphorylation in PC12 cells would be specifically sensitive to propofol.
Discussion
Oxidative stress has been implicated as a potential contributor to the pathogenesis of acute central nervous system injury. After brain injury by ischemic or hemorrhagic stroke or trauma, the increased ROS production may lead to tissue damage via different cellular molecular pathways. ROS can cause damage to cardinal cellular components, such as lipids, proteins, and DNA, resulting in subsequent cell death by necrosis or apoptosis. In the present study, the examined concentrations of H2O2 were in the range of those reached under pathophysiological conditions, such as during transient cerebral ischemia[16]. Propofol has been shown to cause protective action on neurons[7], including antioxidative effects[17], inhibitory effects on lipid peroxidation[18], and direct anti-excitotoxic properties[19]. Other studies showed that propofol had a potential to enhance neurological outcome and decrease the infarct size in experimental animal models of stroke[20,21]. However, no evidence has been reported concerning the direct effect of propofol on neuronal-like PC12 cells and their effect on MAP kinase activity. For the first time, this study showed that propofol protected PC12 cells from H2O2-induced cell death, including apoptosis, by measuring MTT reduction, apoptotic nuclei staining, FACS, and caspase-3 activity. We also observed that propofol specifically inhibited H2O2-induced p38 activation, but not ERK1/2 and JNK activation in PC12 cells.
Caspases are a family of specific cysteine proteases whose activation is critical for the intracellular execution of apoptotic death[22]. Among the 10+ caspases that have been identified, caspase-3 is a common effecter to which several procaspases and caspases converge, and therefore, can serve as an apoptotic marker[23] induced by a variety of stimuli. It has been shown that caspase-3 can be activated by H2O2 exposure as a final effecter in apoptotic death in vitro[24,25]. In agreement with this view, our results showed that H2O2 markedly increased caspase-3 activity in PC12 cells. More-over, we investigated the effect of propofol on the H2O2-induced effect, where we found that propofol inhibited H2O2-induced caspase-3 activation, as shown in Figure 3. However, since the inhibitory effect of 10 µmol/L propofol on H2O2-induced caspase-3 activation was incomplete (Figure 3), the involvement of other mechanisms of propofol protective effects against H2O2-induced PC12 cells death can not be denied. Further studies are required to define the exact role of caspase-3 in the mechanism of H2O2-induced PC12 cell death.
Cellular signaling pathways are regulated by the intracellular redox state of the cell. ROS may play an important role as a second messenger in signal transduction cascades and may lead to the activation of MAP kinases[26]. Previous studies have shown that H2O2 rapidly activated ERK and p38 proteins in PC12 cells, and the mediation of cell death by ROS may be closely related to MAP kinase signaling[27,28]. In line with these studies, we found in the present study that 3 MAP kinases were rapidly and significantly activated by oxidative stress in cultured PC12 cells (Figure 3). The phosphorylation of the 3 MAP kinases almost simultaneously reached their maximum at 10 min, and then the activity of the 3 MAP kinases decreased gradually (data not shown). There-fore, we investigated the effect of propofol on the activity of the 3 MAP kinases at 10 min. We found that propofol suppressed p38 activation, but not JNK nor ERK1/2 phosphorylations (Figure 4), indicating that p38 is specifically sensitive to propofol, which may contribute to its cellular protective effect under oxidative stress. Previous studies showed that the activation of the p38 MAP kinase induced apoptotic death in neuroblastoma cells[29], while inhibitors of the p38 MAP kinases promoted neuronal survival in vitro and reduced cerebral infarction in vivo[30,31]. In line with these studies, we also found that SB203580, a specific p38 inhibitor, attenuated H2O2-induced PC12 cell death and caspase-3 activity (data not shown), which further verified the role of p38 in oxidative stress-mediated cell death.
In conclusion, this study confirmed a neuroprotective effect of propofol, at clinically relevant concentrations, on H2O2-induced PC12 cell death, including apoptosis. It could be assumed that this beneficial effect of propofol on PC12 cell viability is mediated, at least in part, by an inhibition of the p38 pathway, but not that of ERK1/2 or JNK, which are dramatically activated by H2O2. The results of the present study may shed light on the pharmacological basis for the clinical application of some anesthetic compounds, such as propofol in cerebral ischemic and/or reperfusion injury.
References
- Mickalad M, Hans P, Deby-Dupont G, Hoebeke M, Deby C, Lamy M. Propofol reacts with peroxynitrite to form a phenoxyl radical: demonstration by electron spin resonance. Biochem Biophys Res Commun 1998;249:833-7.
- Saito A, Maier CM, Narasimhan P, Nishi T, Song YS, Yu F, et al. Oxidative stress and neuronal death/survival signaling in cerebral ischemia. Mol Neurobiol 2005;31:105-16.
- Ergun R, Akdemir G, Sen S, Tasci A, Ergungor F. Neuroprotective effects of propofol following global cerebral ischemia in rats. Neurosurg Rev 2002;25:95-8.
- Ito H, Watanabe Y, Isshiki A, Uchino H. Neuroprotective properties of propofol and midazolam, but not pentobarbital, on neuronal damage induced by forebrain ischemia, based on the GABA receptors. Acta Anaesthesiol Scand 1999;43:153-62.
- Daskalopoulos R, Korcok J, Farhangkhgoee P, Karmazyn M, Gelb AW, Wilson JX. Propofol protection of sodium-hydrogen exchange activity sustains glutamate uptake during oxidative stress. Anesth Analg 2001;93:1199-204.
- Lionel JV, Benjamin AG, Frederique MM, André LN, Nicolas J, François MG, et al. Neuroprotective effects of propofol in a model of ischemic cortical cell cultures: role of glutamate and its transporters. Anesthesiology 2003;99:368-75.
- Yu BW, Xue QS, Xia M, Wang ZJ, Chen HZ. The influences of propofol on different kinds of brain injuries in rat brain slices. Zhonghua Yi Xue Za Zhi 2003;83:1176-9.
- Christophe M, Nicolas S. Mitochondria: a target for neuropro-tective interventions in cerebral ischemia-reperfusion. Curr Pharm Des 2006;12:739-57.
- Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, DeFranco DB. Oxidative neuronal injury. The dark side of ERK1/2. Eur J Biochem 2004;271:2060-6.
- Kuhn K, Shikhman AR, Lotz M. Role of nitric oxide, reactive oxygen species, and p38 MAP kinase in the regulation of human chondrocyte apoptosis. J Cell Physiol 2003;197:379-87.
- Ballif BA, Blenis J. Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ 2001;12:397-408.
- Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal 2000;12:1-13.
- Kitagawa K, Matsumoto M, Hori M, Yanagihara T. Neuropro-tective effect of apolipoprotein E against ischemia. Ann N Y Acad Sci 2002;977:468-75.
- Spicer Z, Millhorn DE. Oxygen sensing in neuroendocrine cell and other cell types: pheochromocytoma (PC12) cell as an experimental model. Endocr Pathol 2003;14:277-91.
- Lee SJ, Jin Y, Yoon HY, Choi BO, Kim HC, Oh YK, et al. Ciclo-pirox protects mitochondria from hydrogen peroxide toxicity. Br J Pharmacol 2005;145:469-76.
- Hyslop PA, Zhang Z, Pearson DV, Phebus LA. Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vitro. Brain Res 1995;671:181-6.
- Zhang SH, Wang SY, Yao SL. Antioxidative effect of propofol during cardiopulmonary bypass in adults. Acta Pharmacol Sin 2004;25:334-40.
- Lee H, Jang YH, Lee SR. Protective effect of propofol against kainic acid-induced lipid peroxidation in mouse brain homo-genates: comparison with trolox and melatonin. J Neurosurg Anesthesiol 2005;17:144-8.
- Hans P, Bonhomme V, Collette J, Albert A, Moonen G. Propofol protects cultured rat hippocampal neurons against N-methyl-D-aspartate receptor-mediated glutamate toxicity. J Neurosurg Anesthesiol 1994;6:249-53.
- Young Y, Menon DK, Tisavipat N, Matta BF, Jones JG. Propofol neuroprotection in a rat model of ischaemia reperfusion injury. Eur J Anaesthesiol 1997;14:320-6.
- Yamasaki T, Nakakimura K, Matsumoto M, Xiong L, Ishikawa T, Sakabe T. Effects of graded suppression of the EEG with propofol on the neurological outcome following incomplete cerebral ischaemia in rats. Eur J Anaesthesiol 1999;16:320-9.
- Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 2006;13:1423-33.
- Iwai K, Kondo T, Watanabe M, Yabu T, Kitano T, Taguchi Y, et al. Ceramide increases oxidative damage due to inhibition of catalase by caspase-3-dependent proteolysis in HL-60 cell apopto-sis. J Biol Chem 2003;278:9813-22.
- Demelash A, Karlsson JO, Nilsson M, Björkman U. Selenium has a protective role in caspase-3-dependent apoptosis induced by H2O2 in primary cultured pig thyrocytes. Eur J Endocrinol 2004;150:841-9.
- Song JH, Slot AJ, Ryan RW, Ross GM. Dopamine-induced death of PC12 cells is prevented by a substituted tetrahydronaphthalene. Neuropharmacology 2004;46:984-93.
- Eguchi M, Monden K, Miwa N. Role of MAPK phosphorylation in cytoprotection by pro-vitamin C against oxidative stress-induced injuries in cultured cardiomyoblasts and perfused rat heart. J Cell Biochem 2003;90:219-26.
- Spicer Z, Millhorn DE. Oxygen sensing in neuroendocrine cell and other cell types: pheochromocytoma (PC12) cell as an experimental model. Endocr Pathol 2003;14:277-91.
- Suzaki Y, Yoshizumi M, Kagami S, Koyama AH, Taketani Y, Houchi H, et al. Hydrogen peroxide stimulates c-Src-mediated big mitogen-activated protein kinase 1 (BMK1) and the MEF2C signaling pathway in PC12 cell: potential role in cell survival following oxidative insults. J Biol Chem 2002;277:9614-21.
- Natzker S, Heinemann T, Figueroa-Perez S, Schnieders B, Schmidt RR, Sandhoff K, et al. Cis-4-methylsphingosine phosphate induces apoptosis in neuroblastoma cell by opposite effects on p38 and ERK mitogen-activated protein kinases. J Biol Chem 2002;383:1885-94.
- Horstman S, Kahle PJ, Borasio GD. Inhibitors of p38 mitogen-activated protein kinase promote neuronal survival in vitro. J Neurosci Res 1998;52:483-90.
- Piao CS, Kim JB, Han PL, Lee JK. Administration of the p38 MAPK inhibitor SB203580 affords brain protection with a wide therapeutic window against focal ischemic insult. J Neurosci Res 2003;73:537-44.