
Greater stress protein expression enhanced by combined prostaglandin A1 and lithium in a rat model of focal ischemia1
Introduction
Stroke, most commonly caused by the interruption of blood supply to the brain, is one of the leading causes of disability and mortality in adults. Multiple molecular mechanisms, including energy crisis, oxidative stress, calcium overloading, caspase activation, and inflammation have now been suggested as potential contributors to ischemia-induced neural injury[1,2]. Unfortunately, there has been no satisfactory treatment to prevent neural cell death and the long-term neurological deficits in stroke victims.
In previous studies, we have reported that prostaglandin A1 (PGA1) inhibited the excitotoxin-induced apoptosis of striatal neurons in vivo and the rotenone-induced apoptosis of cultured SH-SY5Y cells in vitro[3,4]. Lithium has been extensively used in the treatment of bipolar mood disorder, and recent study has revealed that it is a neuroprotective drug against a variety of insults, such as glutamate-induced excitotoxicity[5], ischemia-induced neural damage, and other neurodegenerative conditions[6–9]. We have found that PGA1 protects neurons in rodent models of focal ischemia[10], and the combination of PGA1 and lithium generates greater neuroprotection under ischemic conditions, possibly through enhancing the expressions of heat shock protein (HSP)70 and glucose response protein 78 (Grp78)[11].
HSP are a group of stress-induced proteins that act as molecular chaperones regulating the formation, folding, and assembly of protein chains and the translocation of newly-formed proteins[12]. Cerebral ischemia of either focal or global type enhances the expression of HSP genes and pro-teins[13–16], which are speculated to be protective proteins. HSP synthesis is controlled by a family of transcription factors, the heat-shock factors (HSF). Ischemia and heat activate HSF-1, which is present in the cytoplasm as an inactive, monomeric form[17]. Trimerization and phosphorylation, as well as nuclear migration of HSF-1, occur under stress conditions. HSF-1 binds to the heat-shock element, which is present in the promoter of the stress response gene and then initiates HSP transcription and synthesis.
Mammalian HSP90 is one of the most abundant cytosolic proteins in eukaryotes, amounting to ~1% of soluble proteins, even in the absence of stress[18,19]. Previous studies found that HSP90 could form a cytosolic complex with apoptosis protease activating factor-1 (Apaf-1) and inhibit cytochrome c-mediated oligomerization of Apaf-1 and the activation of procaspase-9 acting as an anti-apoptotic factor[20].
The upregulation of the expression of HSP70 and Grp78 by prostaglandin A1 (PGA1) and lithium (Li) has been reported[11]. Still, we do not know whether other members of the HSP family are involved in the neuroprotection of combined PGA1 and lithium. The present study was undertaken to explore whether the expressions of other HSP regulating cell death/survival are affected by PGA1 and Li. The effect of PGA1 and Li on Apaf-1, one pro-apoptotic factor regulated by HSP90, was also examined. The results indicated that lithium enhanced the effects of PGA1 on the expressions of HSF-1, heme oxygenase-1 (HO-1), HSP90α, and HSP90β and downregulated Apaf-1.
Materials and methods
Materials Lithium and PGA1 were purchased from Sigma-Aldrich Chemical (St Louis, MO, USA).
Rat permanent middle cerebral artery occlusion model Male Sprague-Dawley rats weighing 280–300 g were purchased from the Center for Experimental Animals of Soochow University (Suzhou, China). The National Institutes of Health (NIH) Guidelines for Care and Use of Laboratory Animals were followed in all animal procedures. The rats were anesthetized with an intraperitoneal injection of 4% choral hydrate (350 mg/kg). Through a ventral midline incision, the right common carotid artery (CCA), external carotid artery, and internal carotid artery (ICA) were isolated and ligated. A 30 mm length of monofilament nylon suture (Φ0.22–0.24 mm), with its tip rounded by heating near a flame, was inserted from the right CCA to the ICA through a small incision in the common carotid artery and then advanced to the Circle of Willis to occlude the origin of the right middle cerebral artery. The suture remained there until the rats were killed[21]. The body temperature of the rats was monitored with a rectal probe and maintained in the range of 37.0±0.5 oC with a heating pad (BME-412A ANIMAL REGULATOR, 308005669; Institute of Biomedical Engineering, Hangzhou, Zhejiang, China) during and after surgery until recovery from anesthesia. Sham-operated rats underwent the same procedures, except for the permanent middle cerebral artery occlusion (pMCAO). Rats showing tremor and seizure after surgery were excluded from further experiments. For the study of the time-course of alterations in the expression of HSF-1, HSP, and Apaf-1, 15 rats were killed at 1, 2, 3, 6, 12, and 24 h after ischemic insult (3 rats in each group). For the study of the effects of PGA1, lithium alone, and a combination of both, 21 rats were divided into 7 groups: sham operated; model; PGA1 33.3 nmol PGA1(L); PGA1 16.5 nmol PGA1(S); lithium 1 mEq/kg; PGA1(L)+lithium; PGA1(S)+lithium.
Lithium and PGA1 treatment For all the experiments, at the indicated times and doses, lithium was administered subcutaneously to the rats for 2 d (1 mEq/kg, once a day) and PGA1 (16.5 or 33 nmol, dissolved in 40% ethanol) was injected into the lateral cerebro-ventricle once 15 min prior to the pMCAO. The control animals received the same quantity of vehicle.
Western blot analysis of HO-1, HSP90α, HSP90β, HSF-1, and Apaf-1 Immunoblotting was performed as previously described[3]. The brain tissues from the ischemic striatum of the right middle cerebral artery territory and the corresponding area of the sham-operated rats were rigorously homogenized; the protein concentrations were determined using a BCA kit (Pierce, Rockford, IL, USA). An aliquot of 50 µg proteins from each sample was separated using 10% SDS-PAGE electrophoresis using constant current. Proteins were subsequently transferred to a nitrocellulose membrane, which was then incubated with 5% skim milk in Tris-buffered saline with 0.1% Tween 20 (TBST) at 4 °C overnight. Afterwards, the membranes were incubated with the mAb to HSF-1 (1:400; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4 °C for 3 h, then with a horseradish peroxidase-conjugated anti-mouse secondary antibody (1:5000; Sigma, USA) at room temperature for 1 h. Immunoreactivity was detected by enhanced chemiluminescent autoradiography (Amersham, Princeton, NJ, USA) in accordance with the manufacturer’s instructions. Between each procedure, the membranes were rinsed 3 times in TBST for 10 min each. The membrane was immediately placed with the protein side up in the film cassette for exposing. The films were used for the final determination. The membranes were reprobed with β-actin (1:5000; Sigma, USA) after striping (TBST with 2% β-mercaptoethanol at 65 °C for 1 h).
The same method was used to detect HO-1, HSP90α, HSP90β, and Apaf-1. For the detection of HO-1, HSP90α, and HSP90β, the first antibodies used to incubate the membranes were rabbit polyclonal IgG against HO-1 (1:200; Stressgen Bioreagents, Voctoria, BC, Canada), rat monoclonal IgG against HSP90α (1:200; Santa Cruz Biotechnology, USA), rat monoclonal IgG against HSP90β (1:200; Santa Cruz Biotechnology, USA), and rabbit polyclonal IgG against Apaf-1 (1:500; Pharmingen, San Jose, CA, USA), and the second antibody was a horseradish peroxidase-conjugated anti-rabbit IgG (1:5000; Sigma, USA) and anti-mouse IgG (1:5000; Sigma, USA).
Statistical analysis Statistical analysis was performed by one-way ANOVA. P<0.05 was considered to be significant.
Results
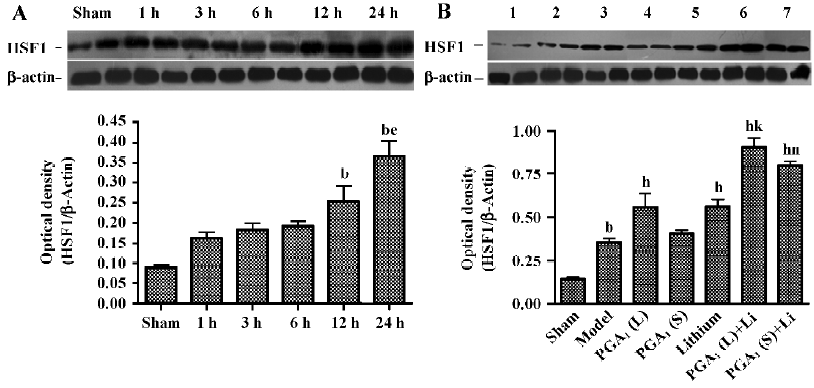
Enhanced expression of HSF-1 by combined PGA1 and lithium The Western blot analysis revealed that the increase in the levels of HSF-1 in the ischemic striatum was detected at 12 h and further elevated 24 h after the onset of ischemia. The levels of HSF-1 were further elevated by pretreatment with PGA1 (33 nmol) and lithium alone. Moreover, the combination of lithium and PGA1 (16.5 and 33 nmol, respectively) elicited a greater increase in HSF-1 levels compared to PGA1 alone (P<0.05; Figure 1).
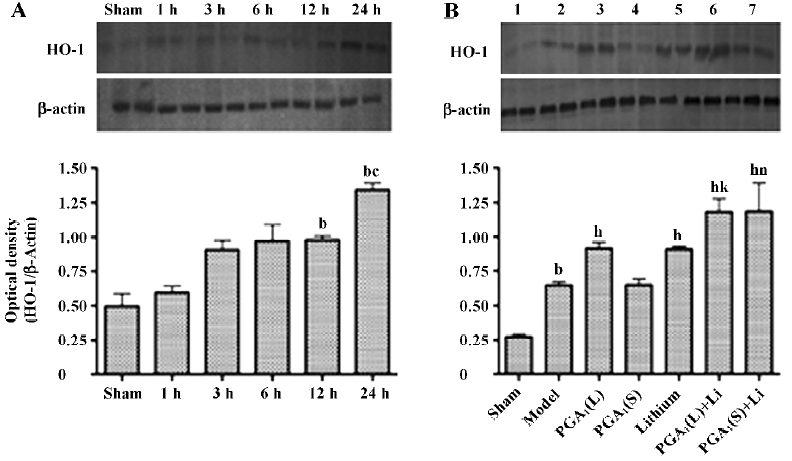
Enhanced expression of HO-1 by combined PGA1 and lithium The elevation of the levels of HO-1 in the ischemic striatum was detected at 12 h and further increased 24 h after the onset of ischemia. The increase in the expression of HO-1 was enhanced by pretreatment with PGA1 (33 nmol) and lithium alone. The combination of lithium and PGA1 (16.5 and 33 nmol, respectively), however, induced a greater increase in HO-1 levels compared to PGA1 alone (P<0.05; Figure 2).
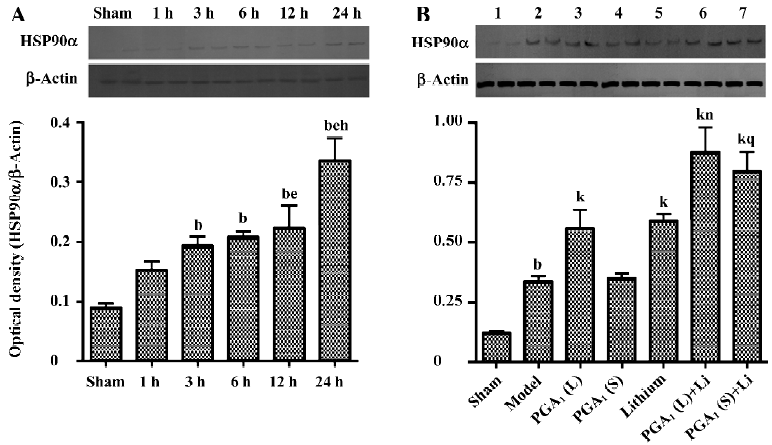
Enhanced expression of HSP90α by combined PGA1 and lithium The upregulation of the expression of HSP90α was detectable at 3 h and was further elevated 24 h after the onset of ischemia. The levels of HSP90α were even further increased by pretreatment with PGA1 (33 nmol) and lithium alone. The combination of lithium and PGA1 (16.5 and 33 nmol, respectively) elicited a larger rise in HSP90α levels compared to PGA1 alone (P<0.05; Figure 3).
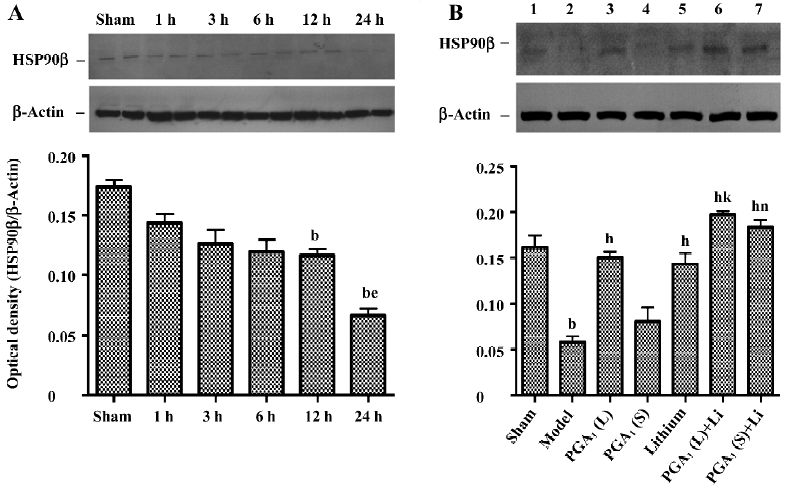
Maintenance of HSP90β expression by combined PGA1 and lithium The downregulation of the expression of HSP90β in the ischemic striatum was detected at 12 h and further descended 24 h after the onset of ischemia. However, it was significantly recovered by pretreatment with PGA1 (33 nmol) and lithium alone. Moreover, the combination of lithium and PGA1 (16.5 and 33 nmol, respectively) elicited a greater rise in HSP90β levels in the ischemic striatum compared to PGA1 alone (P<0.05; Figure 4).
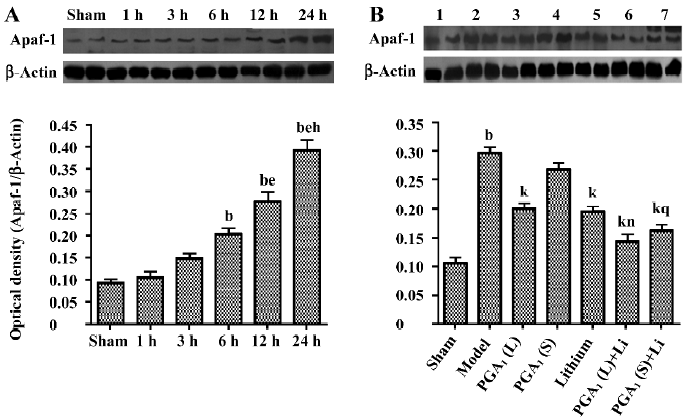
Inhibition of Apaf-1 expression by combined PGA1 and lithium The increased expression of Apaf-1 was detected at 6 h and was further elevated 24 h after the onset of ischemia. Pretreatment with PGA1 (33 nmol) or lithium significantly inhibited Apaf-1 induction. The combination of lithium and PGA1 (16.5 and 33 nmol, respectively) further reduced Apaf-1 levels in the ischemic striatum compared to PGA1 alone (P<0.05; Figure 5).
Discussion
HSF-1 has been shown to regulate the expression of HSP in response to ischemia, hypoxia, heat, stress, or injury[41]. During cerebral ischemia, complex and multiple pathological events occur within neural cells, such as the loss of high energy phosphate esters, disturbances in neurotransmitter metabolism, membrane breakdown, mitochondrial failure, an accumulation of intracellular Ca2+, and the subsequent production of oxygen free radicals involving the arachidonic acid metabolic pathway[42]. Although the detailed mechanisms remain to be determined, complex pathological events within ischemic neuronal cells disturb protein metabolism and break down cellular structures, thus possibly producing substrates for the molecular chaperones and activating HSF-1. Conditions that increase the level of oxygen free radicals have also been reported to induce the stress proteins[43]. Hypoxia also activates HSF-1 and the HSF-mediated transcription of HSP70 in myogenic cells[44]. Mosser et al reported that the concentrations of calcium and hydrogen ions, which affect protein conformation, directly activate HSF in vitro[45]. Recent studies have reveled that antiproliferative prostaglandins also activate HSF, and extracellular exposure to arachidonic acid activates HSF-1 and heat shock gene transcription[46,47]. It has been reported recently that cerebral ischemia or heat shock could activate HSF-1[48]. Our current findings demonstrate that pMCAO enhanced the expression of HSF-1 in the ischemic striatum, and pre-ischemic PGA1 and lithium treatments further enhanced the induction of the HSF-1 level in the ischemic striatum. This suggests that the enhanced expression of HSP by PGA1 and lithium is mediated at the transcriptional level through HSF-1.
HO-1, a HO family member, is a 32 kDa HSP (HSP-32), which is inducible by a variety of stress factors, including oxidative stress, exposure to heavy metals, heat, focal cerebral ischemia, and traumatic brain injury[22–24]. In the human central nervous system, HO-1 expression was detected in brain tumors and in neurodegenerative diseases including Pick’s disease, Alzheimer’s disease, Parkinson’s disease, and cerebral ischemia[25–27]. It should be noted that the functional role of HO-1 is still unclear. There are several indications of the protective function of HO-1. Oxidative stress has been proposed to be a contributing factor in the pathophysiology of ischemia/reperfusion[28]. The products of heme degradation, bilirubin, and biliverdin were shown to be protective because of their antioxidant activity[29,30]. HO-1 was shown to protect vessels against heme and hemoglobin-mediated injury[31], to protect kidney against ischemia/perfusion-induced injury[32] and to reduce hyperoxia-induced lung injury in rats[33]. The increased expression of the HO-1 protein following transient global and permanent focal ischemia may reflect an elevation of antioxidant defense mechanisms as a response to ischemia-induced oxidative stress. In our ischemic model, the HO-1 level was maximally increased at 24 h post-insult in the ischemic striatum, when the brain area suffered severe damage after pMACO. Moreover, pre-ischemic PGA1 and lithium treatments robustly enhanced the induction of the HO-1 level in the ischemic striatum.
HSP90 is a highly conserved and essential stress protein for the viability of eukaryotic cells. It is not only abundantly expressed in cells, but also significantly induced after stress[34]. In eukaryotes, HSP90 has dual chaperone functions. They are involved in the conformational maturation of signal transduction molecules (for example, nuclear hormone receptors and kinases) and in the cellular stress response[18]. The finding that HSP90 is induced in response to diverse apoptotic stimuli, such as UV, sodium arsenite and cerebral ischemia, has supported its involvement in cell survival[35–37]. Recently, it was reported that HSP90 could form a cytosolic complex with Apaf-1 and inhibit cytochrome c-mediated oligomerization of Apaf-1 and the activation of procaspase-9[20]. It also could stabilize the receptor-interacting protein, a major anti-apoptotic adaptor, to activate anti-apoptotic pathways through NF-κB and mitogen-activated protein kinases (MAPK)[38]. In addition, Akt is a substrate of HSP90 and forms a complex with HSP90. The inhibition of Akt–HSP90 binding results in Akt dephosphorylation, a decrease in Akt kinase activity, and the induction of apoptosis[39]. In the present study, although HSP90α expression was enhanced significantly by pMCAO, HSP90β expression was significantly decreased in the ischemic models. Pre-ischemic PGA1 and lithium treatment robustly enhanced the induction of HSP90α levels and maintained the expression of HSP90β in the ischemic striatum. Our findings in changes in HSP90β expression is consistent with those reported by Sun et al, in which HSP90β descended significantly in the ischemic/reperfusion model, but was maintained by the treatment of acupuncture[40]. This may contribute to PGA1 and lithium’s protection against cerebral ischemia in our study.
Taken together, pretreatment with PGA1, lithium, and a combination of both just before cerebral ischemia, significantly enhanced the expressions of HO-1, HSP90α, and HSF-1, and maintained HSP90β, but decreased the expression of Apaf-1 in the ischemic striatum. This is the first in vivo study showing that pretreatment with PGA1, lithium, and a combination of both could activate heat shock gene transcription, suggesting that the combined treatment of PGA1 and lithium may be considered for the clinical testing of PGA1 and lithium against ischemic neuronal damage.
References
- Stoll G, Jander S, Schroeter SM. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol 1998;56:149-71.
- White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, et al. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 2000;179:1-33.
- Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, Chase TN. Nuclear factor kappaB nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor-mediated apoptosis in rat striatum. J Neurosci 1999;19:4023-33.
- Wang X, Qin ZH, Leng Y, Wang Y, Jin X, Chase TN, et al. Prostaglandin A1 inhibits rotenone-induced apoptosis in SH-SY5Y cells. J Neurochem 2002;83:1094-102.
- Chen RW, Qin ZH, Ren M, Kanai H, Chalecka-Franaszek E, Leeds P, et al. Regulation of c-Jun N-terminal kinase, p38 kinase and AP-1 DNA binding in cultured brain neurons: roles in glutamate excitotoxicity and lithium neuroprotection. J Neurochem 2003;84:566-75.
- Nonaka S, Chuang DM. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport 1998;9:2081-4.
- Wei H, Qin ZH, Senatorov VV, Wei W, Wang Y, Qian Y, et al. Lithium supresses excitotoxicity-induced striatal lesions in a rat model of Huntington’s disease. Neuroscience 2001;106:603-12.
- Ren M, Senatotov VV, Chen RW, Chuang DM. Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc Natl Acad Sci USA 2003;100:6210-5.
- Xu J, Culman J, Blume A, Brecht S, Gohlke P. Chronic treatment with a low dose of lithium protects the brain against ischemic injury by reducing apoptotic death. Stroke 2003;34:1287-92.
- Zhang HL, Huang ZH, Zhu Y, Liang ZQ, Han R, Wang X, et al. Neuroprotective effects of prostaglandin A1 in animal models of focal ischemia. Brain Res 2005;1039:203-6.
- Xu XH, Zhang HL, Han R, Gu ZL, Qin ZH. Enhancement of neuroprotection and heat shock protein induction by combined prostaglandin A1 and lithium in rodent models of focal ischemia. Brain Res 2006;1102:154-62.
- Hendrick JP, Hartl FU. Molecular chaperone functions of heat-shock proteins. Annu Rev Biochem 1993;62:349-84.
- Fu R, Zhao ZQ, Zhao HY, Zhao JS, Zhu XL. Expression of heme oxygenase-1 protein and messenger RNA in permanent cerebral ischemia in rats. Neurological Res 2006;28:38-45.
- Fredduzzi S, Mariucci G, Tantucci M, Ambrosini MV. Generalized induction of 72-kDa heat-shock protein after transient focal ischemia in rat brain. Exp Brain Res 2001;136:19-24.
- Izaki K, Kinouchi H, Watanabe K, Owada Y, Okubo A, Itoh H, et al. Induction of mitochondrial heat shock protein 60 and 10 mRNAs following transient focal cerebral ischemia in the rat. Brain Res Mol Brain Res 2001;88:14-25.
- Majda BT, Meloni BP, Rixon N, Knuckey NW. Suppression subtraction hybridization and northern analysis reveal upregula-tion of heat shock, trkB, and sodium calcium exchanger genes following global cerebral ischemia in the rat. Brain Res Mol Brain Res 2001;93:173-9.
- Takaki E, Fujimoto M, Sugahara K, Nakahari T, Yonemura S, Tanaka Y, et al. Maintenance of olfactory neurogenesis requires HSF-1, a major heat shock transcription factor in mice. J Biol Chem 2006;281:4931-7.
- Csermely P, Schnaider T, Soti C, Prohaszka Z, Nardai G. The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacol Ther 1998;79:129-68.
- Minami Y, Kawasaki H, Miyata Y, Suzuki K, Yahara I. Analysis of native forms and isoform compositions of the mouse 90-kDa heat shock protein, HSP90. J Biol Chem 1991;266:10099-103.
- Pandey P, Saleh A, Nakazawa A, Kumar S, Srinivasula SM, Kumar V, et al. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J 2000;19:4310-22.
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989;20:84-91.
- Dwyer BE, Nishimura RN, Lu SY, Alcaraz A. Transient induction of heme oxygenase after cortical stab wound injury. Mol Brain Res 1996;38:251-9.
- Siesjo BK, Katsura K, Kristian T. The biological basis of cerebral ischemic damage. J Neurosurg Anesthesiol 1995;7:47-52.
- Takeda A, Kimpara T, Onodera H, Itoyama Y, Shibahara S, Kogure K. Regional difference in induction of heme oxygenase-1 protein following rat transient forebrain ischemia. Neurosci Lett 1996;205:169-72.
- Hara ED, Takahashi K, Tominaga T, Kumabe T, Kayama T, Suzuki H, et al. Expression of heme oxygenase and inducible nitric oxide synthase mRNA in human brain tumours. Biochem Biophys Res Commun 1996;224:153-8.
- Premkumar DR, Smith MA, Richey PL, Petersen RB, Castellani R, Kutty RK, et al. Induction of heme oxygenase-1 mRNA and protein in neocortex and cerebral vessels in Alzheimer’s disease. J Neurochem 1995;65:1399-402.
- Geddes JW, Pettigrew LC, Holtz ML, Craddock SD, Maines MD. Permanent focal and transient global cerebral ischemia increase glial and neuronal expression of heme oxygenase-1, but not heme oxygenase-2, protein in rat brain. Neurosci Lett 1996;210:205-8.
- Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia . J Neurosci 2005; 25: 10 321–35.
- Elbirt KK, Bonkovsky HL. Heme oxygenase: recent advances in understanding its regulation and role. Proc Assoc Am Physicians 1999;111:438-47.
- Suttner DM, Dennery PA. Reversal of HO-1 related cytoprotec-tion with increased expression is due to reactive iron. FASEB J 1999;13:1800-9.
- Abraham NG, Lavrosvky Y, Schwartzman ML, Stoltz RA, Levere RD, Gerritsen ME, et al. Transfection of the human hemeoxy-genase gene into rabbit coronary microvessel endothelial cells: protective effect against heme and hemoglobin toxicity. Proc Natl Acad Sci USA 1995;92:6798-802.
- Maines MD. Theheme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 1997;37:517-54.
- Otterbein LE, Kolls JK, Mantell LL, Cook JL, Alam J, Choi AMK. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J Clin Invest 1999;103:1047-54.
- Jakob U, Buchner J. Assisting spontaneity: the role of Hsp90 and small Hsps as molecular chaperones. Trends Biochem Sci 1994;19:205-11.
- Parsell DA, Lindquist S. The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu Rev Genet 1993;27:437-96.
- Ali A, Krone PH, Pearson DS, Heikkila JJ. Evaluation of stress-inducible hsp90 gene expression as potential molecular biomarker in Xenopus laevis. Cell Stress Chaperones 1996;1:62-9.
- Yang SH, Liu R, Wen Y, Perez E, Cutright J, Brun-Zinkernagel AM, et al. Neuroendocrine mechanism for tolerance to cerebral ischemia-reperfusion injury in male rats. J Neurobiol 2005;62:341-51.
- Lewis J, Devin A, Miller A, Lin Y, Rodriguez Y, Neckers L, . Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J Biol Chem 2000; 275: 10 519–26.
- Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci USA 2000; 97: 10 832–7.
- Sun N, Shi J, Chen L, Liu X, Guan X. Influence of electro-acupuncture on the mRNA of heat shock protein 70 and 90 in brain after cerebral ischemia/reperfusion of rats. J Huazhong Univ Sci Tech Med Sci 2003;23:112-5.
- Bergeron M, Mivechi NF, Giaccia AJ, Giffard RG. Mechanism of heat shock protein 72 induction in primary cultured astrocytes after oxygen-glucose deprivation. Neurol Res 1996;18:64-72.
- Choi DW. Cerebral hypoxia: Some new approaches and unanswered questions. J Neurosci 1990;10:2493-501.
- Sciandra JJ, Subjeck JR, Hughes CS. Induction of glucose-regulated proteins during anacrobic exposure and of heat-shock proteins after reoxygenation. Proc Natl Acad Sci USA 1984;81:4843-7.
- Mestril R, Chi SH, Sayen MR, Dillmann WH. Isolation of a novel inducible rat heat-shock protein (HSP70) gene and its expression during ischemia/hypoxia and heat shock. Biochem J 1994;298:561-9.
- Mosser DD, Kotzbauer PT, Sarge KD, Morimoto RI. In vitro activation of heat shock transcription factor DNA-binding by calcium and biochemical conditions that affect protein confor-mation. Proc Natl Acad Sci USA 1990;87:3748-52.
- Amici C, Sistonen L, Santoro MG, Morinoto RI. Antiproliferative prostaglandins activate heat shock transcription factor. Proc Natl Acad Sci USA 1992;89:6277-81.
- Jurivich DA, Sistonen L, Sarge KD, Morinoto RI. Arachidonate is a potent modulator of human heat shock gene transcription. Proc Natl Acad Sci USA 1994;91:2280-4.
- Higashi T, Nakai A, Uemura Y, Kikuchi H, Nagata K. Activation of heat shock factor 1 in rat brain during cerebral ischemia or after heat shock. Brain Res Mol Brain Res 1995;34:262-70.