
Effect of TGF-β/Smad signaling pathway on lung myofibroblast differentiation1
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic diffuse interstitial lung disease of unknown cause, characterized by progressive fibrosis of lung parenchyma[1,2]. A major process in fibrosis development is the differentiation of fibroblastic cells into myofibroblasts which express α-smooth muscle actin (α-SMA). Since myofibroblasts are virtually absent in normal lungs, their differentiation is one of the decisive events in the pathogenesis of progressive lung fibrosis[3–5] and are largely responsible for the accumulation of extracellular matrix (ECM). Therefore, it is very important to identify the signaling pathway responsible for myofibro-blasts differentiation and accumulation in the lung tissue.
Transforming growth factor (TGF)-β1 has been widely recognized as a key fibrogenic cytokine and has been demonstrated to activate fibroblasts differentiation into myofibroblasts in vitro and in vivo[6,7], and stimulate ECM production[8]. The major signaling pathway of TGF-β1 is through its transmembrane receptor serine/threonine kinases and activate the cytoplasmic Smad proteins[9,10]. Among those, Smad2 and Smad3 are receptor-activated Smad proteins (R-Smad) phosphorylated by TGF-β receptor (TβR)type I, and subsequently form a heteromeric complex with Co-Smad (Smad4), respectively[9,10]. Such complexes subsequently translocate into the nuclear to regulate the expression of target genes. In addition to these positively acting Smad, Smad7 antagonizes TGF-β1 signal by interacting with the receptor complex, and preventing phosphorylation of R-Smad[9,10]. All Smad proteins have conserved amino-terminal MH1 and carboxy-terminal MH2 domains. The MH1 domain is important for mediating the DNA-binding activity of Smad. The MH2 has a Serine-Serine-X-Serine (SSXS)conserved domain which is phosphorylated by TβR-I[11]. Although Smad2 is structurally highly similar to Smad3, and both Smad2 and Smad3 are phosphorylated directly by the TβR-I, they do not share similar DNA-binding activity. This notable difference in transcriptional activation by Smad2 and Smad3 involves an insertion of 30 amino acids in the N-terminal MH1 domain of Smad2[12]. Studies have found that the targeted deletion of Smad2 or Smad3 genes in mice has revealed distinct developmental roles for these closely related Smad[13,14]. Studies about fibroblasts derived from embryos null for either Smad3 or Smad2 also revealed that TGF-β-regulated genes depend on either Smad2 or Smad3 or both[15]. The role of individual Smad signaling in myofibro-blasts differentiation is still unclear.
TGF-β1 plays important roles in a variety of developmental and pathological processes[11]. It is obvious that any attempt to reverse fibrosis by using neutralizing antibodies to block the whole function of TGF-β1 will result in disaster, therefore, it will be more applicable that we select a down-stream point of this pathway to more specifically block the process of fibrosis.
Materials and methods
Cell culture Human fetal lung fibroblasts were purchased from Cell Center, Chinese Academy of Medical Sciences (Beijing, China) and Peking Union Medical College (China) The cells were grown in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum (FBS), antibiotics (100 U/mL penicillin and 100 mg/mL streptomycin) and 25 mmol/L HEPES under conditions of humidified 5% CO2/95% air at 37 °C.
Antibodies and reagents Active human recombinant TGF-β1 was purchased from Peprotech (London, UK); mouse monoclonal anti-α-SMA antibody from DAKO (Glostrup, Denmark); mouse monoclonal anti-flag M2 antibody from Sigma (St Louis, Missouri, USA).
Plasmids: The human α-SMA promoter gene was cloned by PCR from human genomic DNA with primers 5'-GAATT-CGAGACGAGATTTGGG-3'(–895/–875) and 5'-GTGGTGTT-CAGGGAAGCTGA-3' (+9/–11). It was inserted into vector pGal3-basic (Promega,Madison,WI,USA) at the MluI/XhoI site to form the α-SMA-luciferase(Luc) fusion plasmid p895-Luc. pFlag-Smad7, pFlagSmad3, pFlagSmad3mut and pFlagSmad2 were expression vectors for Smad7, Smad3, Smad3mut and Smad2, respectively. These vectors were kindly supplied by Dr Dijke-Peter TEN (Ludwig Institute for Cancer Research, Uppsala, Sweden), Dr Harvey F LODISH (Depart-ment of Chemistry and Biochemistry, University of Boulder, Colorado, USA) and Dr Joan MASSAGUE (Sloan Kettering Memorial Cancer Center, New York, NY, USA). Smad3mut is a dominant negative mutant with 3 C-terminal serine phosphorylation sites changed to alanines[16]. pFlag-Smad2 was used as the template for obtaining Smad2mut by PCR and also has 3 C-terminal serine phosphorylation sites changed to alanines. Smad2mut was subcloned in vector pFlag-Smad2 to form the pFlagSmad2mut. pSV-β-galactosidase vector was used as the standard for transfection efficiency.
Transfection of cells and reporter gene assay Transient cotransfections were performed using the FuGENETM 6.0 reagent (Roche Molecular Biochemicals, Indianapolis, IN, USA) according to the manufacturer’s instructions. Supercoiled DNA was isolated with an endotoxin-free Qiagen column kit (Qiagen, Inc Valencia, CA). Unless otherwise indicated, cells were seeded in 24-well plates at a density of 2.5×104 cells per well in medium containing 10% FBS. Twenty-four hours later, the indicated DNA plasmids were transfected in medium containing 0.1% FBS. The total amount of plasmid DNA within each experiment was kept constant by the addition of appropriate empty vectors. For reporter gene assay, pSV-β-galactosidase control vector was cotransfected for normalization. Eight hours following transfection, cells were incubated with TGF-β1. After 24 h the cells were harvested, and luciferase activity and β-galactosidase activity was measured by using the luciferase assay system and β-galactosidase assay kit from Promega, respectively. There were 3 independent experiments and triplicates every time for each plasmid. The results are expressed as relative luciferase activity (RLA). The RLA of the control group is 1.
Western blot analysis Transient cotransfections were performed as above. Eight hours following transfection, the cells were incubated with TGF-β1 for 3 d, then whole cell lysates were prepared with lysis buffer containing protease inhibitor. In the animal experiment, lung tissues were homogenized and proteins were extracted in lysis buffer. The protein concentrations were measured by using protein assay reagent kit (Pierce, Rockford, IL, USA). Fifty mg cell protein and 50 mg tissue protein were electrophoresed on 12% SDS-polyacrylamide gel, and wet transferred onto polyvinylidene difluoride membrane, respectively. The membranes containing the cell proteins were incubated overnight at 4oC with anti-α-SMA antibody, anti-Smad4 antibody and anti-Flag M2 antibody, respectively. The membranes containing tissue proteins were incubated with anti-α-SMA antibody. Then the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Signals were visualized with chemiluminescence reagents (Pierce, Rockford, IL,USA). Quantification of the bands was performed by using densitometric analysis software-Quantity One (Bio-Rad, Hercules, CA, USA)
Animal model To further confirm the role of Smad3 on myofibroblasts differentiation in vivo, we used Smad3 knockout mice, which were bred from heterozygous mice of a targeted disruption of exon8 of the Smad3 gene, kindly supplied by Dr Chu-xia DENG (NIH, Washington, USA). The genotype of the mice was determined by PCR analysis of tail DNA. The mice were maintained on standard chow and allowed access to food and water ad libitum. The mice (8–10 weeks of age) were randomly selected for either bleomycin or saline vehicle control treatment. The average body weight for wild-type mice was ~27 g and for the Smad3 knockout mice, ~17 g. Administration of bleomycin (Nippon Kayaku Co Ltd, Tokyo, Japan) or saline vehicle was performed by a constant subcutaneous infusion through a micro-osmotic pump (model 1007D; Alza, Palo Alto, CA, USA) from d 0–7. In the mice anesthetized with pentobarbital sodium (~50 mg/g, ip), the mini pump loaded with bleomycin (0.15 mg/g mouse body weight dissolved in saline) was implanted subcutaneously on the back of the mice, slightly posterior to the scapulae[17]. The mice were killed after 4 weeks, and the pumps were examined to ensure that they had delivered the entire dosage in each mouse. No significant changes of the body weight were observed after 4 weeks’ treatment. The lungs were removed for protein extraction, measurements of hydroxyproline content, or fixation for histological analysis.
Histology and immunohistochemistry The mouse lungs were fixed by perfusion with 10% formaldehyde before routine processing and paraffin embedding. Serial sections, 4 µm thick, were prepared and stained with hematoxylin and eosin (H&E) for histological examination. Alternatively, lung sections were processed for Masson’s trichrome stain for collagen distribution. Lung tissue sections were deparaf-finized, and endogenous peroxidase was blocked. Sections were treated with blocking goat serum for 15 min and incubated overnight with anti-α-SMA antibody (1:100), then with biotinylated link secondary antibody and peroxidase-labeled streptavidin followed by a diaminobenzidine revelation, and a counterstaining with Mayer’s hematoxylin.
Hydroxyproline quantification of lung tissue The total hydroxyproline content of 50 mg lung tissue was measured as an assessment of lung collagen content. The lung tissues were homogenized in ice-cold PBS, hydrolyzed for 3 h at 130 oC in 4 mL 6 mol/L HCl. After NaOH neutralization, and adjusted solution, the pH level was between 6 and 8. Hydroxyproline content was determined with a color-based reaction as described by Stegemann and Stalde[18]. Concentrations of unknown hydroxyproline were quantified according to the standard curve for hydroxyproline.
Statistical analysis Data were expressed as mean±SD. The differences in mean values were analyzed by one-way ANOVA test and post hoc analysis with the Bonferroni method with SPSS 11.0 software (SPSS Inc, Chicago, Illinois, USA). P<0.05 was considered statistically significant.
Results
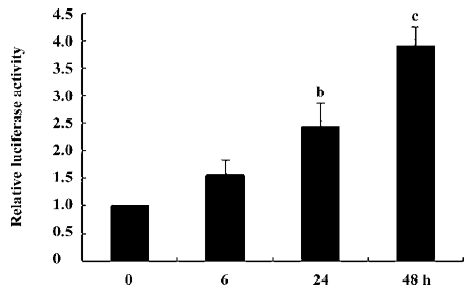
Analysis of human α-SMA promoter-driven Luc activity in human lung fibroblasts To study α-SMA expression at the transcriptional level, a ~904 bp human α-SMA promoter was fused with the luciferase reporter gene to form an α-SMA-Luc fusion plasmid called p895-Luc. We first measured α-SMA promoter-driven Luc activity in human lung fibroblasts. As shown in Figure 1, 5 ng/mL TGF-β1 can induce α-SMA promoter-driven Luc activity in a time-dependent manner. Compared to the cells without TGF-β1 treatment, α-SMA promoter-driven Luc activity increased about 2.44-fold (P=0.01) in the TGF-β1-treated 24 h cells and 3.91-fold (P=0.000) in the TGF-β1-treated 48 h cells. This indicates that the construction of the plasmid p895-Luc is fully functional and can be used in our further studies.
Effect of Smad protein on α-SMA expression in human lung fibroblasts We then examined the possible role of individual Smad protein on α-SMA expression at the transcriptional level and at protein translational level in human lung fibroblasts. To confirm the function of each Smad in TGF-β1-induced human α-SMA expression, we employed loss-of-function mutations Smad2mut or Smad3mut, in which the replacement of 3 C-terminal serine residues with alanine interferes with Smad2 or Smad3 phosphorylation by the activated TGF-β1 receptor. If the Smad cascade is responsible for the upregulation of α-SMA gene expression by TGF-β1 receptor activation, then adding plasmids containing a dominant-negative Smad2mut, dominant-negative Smad3mut, or inhibitory Smad7 should reverse α-SMA gene expression induced by TGF-β1.
Smad plasmid transfection did not affect Smad4 expression in human lung fibroblasts Smad4 is essential for the Smad3/Smad2 signaling pathway of TGF-β1. First, we confirmed that the overexpression of the Smad3, Smad3mut, Smad2 or Smad2mut does not affect Smad4 basic expression. As shown in Figure 2, Smad4 protein was still present after the cells were transfected with Smad3, Smad3mut, Smad2, or Smad2mut expression plasmids.

Smad2 did not affect α-SMA expression in human lung fibroblasts As shown in Figure 3A, when p895-Luc was cotransfected with either the Smad2-expressing plasmid or Smad2mut-expressing plasmid into the fibroblasts, both Smad2 and Smad2mut overexpression did not affect basal and TGF-β1-induced α-SMA promoter activity. Western blot analysis also showed that transient overexpression of Smad2 and Smad2mut had no effect on TGF-β1-induced human α-SMA protein expression (Figure 3B).

Smad3 upregulated α-SMA expression in human lung fibroblasts When p895-Luc was cotransfected with either the Smad3-expressing plasmid or Smad3mut-expressing plasmid into fibroblasts, as shown in Figure 4A, although Smad3 and Smad3mut by themselves had no effect on basal α-SMA promoter activity, overexpression Smad3 was able to stimulate TGF-β1-induced promoter activity in a dose-dependent manner, Smad3-expressing plasmid 1.0 µg could significantly increased TGF-β1-induced α-SMA promoter activity (P<0.01). Overexpression of Smad3mut was able to inhibit TGF-β1-induced α-SMA promoter activity in a dose-dependent manner. Smad3mut expressing plasmid 1.0 µg could significantly inhibit TGF-β1-induced α-SMA promoter activity (P<0.05). Western blot analysis also showed that the transient overexpression of Smad3 increased TGF-β1-induced human α-SMA protein expression, and overexpression of Smad3mut inhibited TGF-β1-induced human α-SMA protein expression (Figure 4B).
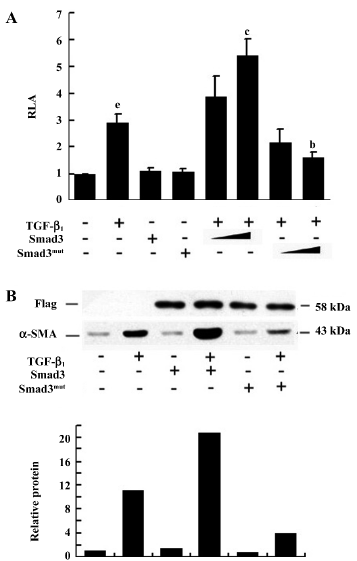
Smad7 downregulated α-SMA expression in human lung fibroblasts Smad7 forms stable interaction with the activated TGF-β receptor and thus blocks ligand-induced Smad3 phosphorylation. As shown in Figure 5A, transient overexpression of Smad7 in fibroblasts abrogated the TGF-β1-induced α-SMA promoter activity in a dose-dependent manner. 1.0 µg Smad7 expressing plasmid could significantly inhibit TGF-β1-induced α-SMA promoter activity (P<0.05). Western blot analysis also showed that the transient overexpression of Smad7 prevented the TGF-β1-induced α-SMA protein expression (Figure 5B). This indicated that the Smad7 signaling pathway was responsible for the inhibitory response elicited by TGF-β1.

Differentiated myofibroblasts are significantly reduced in the lung tissue of Smad3 knockout mice treated by bleomycin To further ascertain the effect of the Smad3 pathway in vivo, we examined myofibroblast differentiation in bleomycin-induced fibrotic lesions in wild-type mouse and Smad3 knockout mouse.
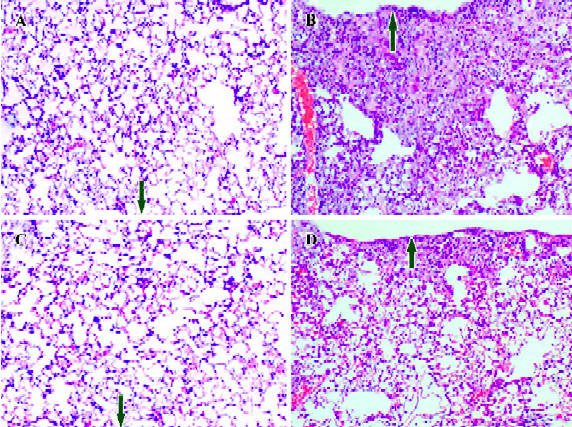
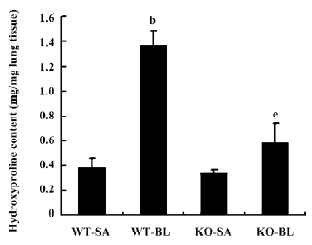
First, histological examination demonstrated that both wild-type and Smad3 knockout mice treated with saline vehicle demonstrated a well-alveolized normal histology (Figure 6A, 6C). Fibrotic lesions induced by bleomycin in wild-type mouse primarily distributed subpleural regions with thickened interalveolar septa (Figure 6B). However, when Smad3 knockout mice were given bleomycin treatment, fibrotic lesions were much less severe in the subpleural regions, and only a slight degree of interstitial fibrogenesis was detected (Figure 6D). The hydroxyproline content of lung tissue was determined to quantify the amount of collagen. As shown in Figure 7, hydroxyproline content significantly increased in the bleomycin-treated wild-type mice vs saline control mice (1.37±0.12 vs 0.38±0.08 µg/mg lung tissue; P<0.05). Hydroxyproline content was much less in the bleomycin-treated Smad3 knockout mice than in the bleomycin-treated wild-type mice (0.68±0.16 vs 1.37±0.12 µg/mg lung tissue; P<0.05). These results show that loss of Smad3 can attenuate bleomycin-induced fibrotic lesions in mice.
Second, we examined the distribution of collagen deposition by Masson’s trichrome staining and distribution of myofibroblasts by immunohistological stain using α-SMA antibodies. As shown in Figure 8A, collagen deposition was primarily distributed in the subpleural regions. There were many differentiated myofibroblasts (Figure 8C) in collagen deposition regions in the bleomycin-treated wild-type mice. However, in the bleomycin-treated Smad3 knockout mice, collagen deposition (Figure 8B) and the number of myofibroblasts (Figure 8D) were reduced.

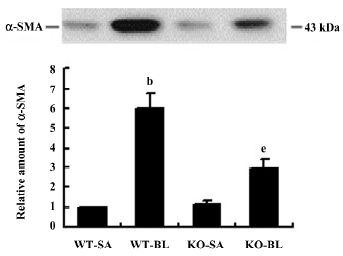
Next we quantified the expression of α-SMA in lung tissue by Western blotting. As shown in Figure 9, the expression of α-SMA was lower in bleomycin-treated Smad3 knockout mice than that in bleomycin-treated wild-type mice (P<0.05). However, this decrease did not drop down to the basic level, compared with the saline-treated wild-type mice.
Discussion
Interstitial myofibroblasts are identified as a key participant in abnormal remodeling and progressive lung fibrosis in IPF[3]. However, relatively little is known about the underlying mechanisms that regulate myofibroblasts differen-tiation. TGF-β1 has been widely recognized as a key fibro-genic cytokine. Extracellular blockade of TGF-β1 signaling, involving interference at the level of the ligand or of its receptor, could ameliorate the fibrotic process in animal model of lung fibrosis[19,20,21], but this is a non-specific blockade and will result in many unacceptable side effects. The targeting of individual intracellular mediators could permit selective blockade of pathological TGF-β1 responses such as fibrosis, without affecting other physiologically important TGF-β1 responses. Therefore, the aim of the current study was to determine the role of individual TGF-β1/Smad signal proteins in mediating α-SMA gene expression, which is the well-known key marker of myofibroblasts differentiation.
It is also known that Smad signal transduction pathways are crucial in mediating several TGF-β1 response in fibro-blasts, such as collagen[22,23], tissue inhibitor of metallopro-teinase-1(TIMP-1)[24], and plasminogen activator inhibitor-1(PAI-1)[25,26]. These transcriptional responses appear to be mediated predominantly through Smad3. In the present study, we demonstrated that it is Smad3, not Smad2, that functions as the major mediator for TGF-β1 signaling in activating human α-SMA gene expression in vitro. This result was also confirmed by a bleomycin-treated Smad3 knockout mice fibrosis model. Hu et al[27] found that the regulation of both basal and TGF-β1-induced rat α-SMA promoter activity is involved in Smad3, but our study demonstrated Smad3 and Smad3mut fail to affect the basal activities of the human SMA promoter. Some investigators also have shown that Smad 2 is capable of activating a TGF-β1-responsive mouse vascular smooth muscle α-actin promoter just as well as Smad 3. The reason for the different results is that the transcriptional regulation of the α-SMA gene is complex and likely to be tissue- and cell-specific[28,29]. There are different nuclear transcription factors which take part in the regulation of α-SMA gene expression in different strain cells and different binding sites for Smad transcriptional complex binding within the α-SMA promoter gene[30]. So the human promoter behaves differently from the mouse promoter in this regard. These need further investigation. This is so far the first report that shows the unique role of Smad3 in the regulation of human α-SMA promoter activity.
It is very clear that both Smad2 and Smad3 are involved in the TGF-β1 signaling pathway, even though they share 92% homology in their amino acid sequence; extensive study has shown that Smad2 and Smad3 have different functions. Smad2 knockout mice are embryonic lethal because of a failure to develop mesoderm[14]. On the other hand, Smad3 knockout mice are viable, although with limb malformations and a defect in immune functions[13]. The studies using Smad2 and Smad3-deficient fibroblasts showed the expressions of both α2 (I) procollagen and matrix metalloproteinase-1(MMP-1)are dependent on Smad3[23,31], and the gelatinase expression is dependent on Smad2[15]. In some instances, such distinct roles can be explained by the lower affinity of Smad2 in DNA binding due to an insertion of 30 amino acids in the N-terminal MH1 domain[12]. Whether there is a factor to differentiate the function of Smad2 from Smad3 in α-SMA gene regulation remains to be determined.
Our study also showed that TGF-β1-induced α-SMA gene expression did not drop down to the basic level when exogenous Smad7 and Smad3mut were added into the fibroblasts. Furthermore, Smad3-deficient mice treated with bleomycin displayed an attenuated pulmonary fibrosis, but the α-SMA level was higher than that of the saline control group. There are 3 explanations for this result. First, a multiplicity of downstream pathways have been shown to be activated by TGF-β1[32], and they form a complex signaling network with extensive cross-talk, such as c-jun-NH2-terminal kinase-dependent pathway which mediates phenotypic modulation of human lung fibroblasts to myofibroblasts induced by TGF-β1[6]. Other signaling pathways may potentially contribute to induce α-SMA gene expression when Smad3 signaling is disrupted. The crosstalk between Smad-dependent and Smad-independent pathways is yet unknown. Second, at whole body level, in addition to TGF-β1, multiple growth factors, cytokines and chemokines have also been implicated in the pathogenesis of fibrosis[33]. Recently, a novel molecule, FIZZ1 (found in inflammatory zones;also known as RELM-α or resistin-like molecule-α) was found to be highly expressed in the bleomycin-induced lung fibrosis model and could activate fibroblast and myofibroblast differentiation independent of TGF-β1[34]. Third, because the origin of myofibroblasts is unclear, cells other than lung interstitial fibroblast, such as circulation fibrocytes and bone marrow-derived multipotent cells could differentiate into myofibroblasts[35,36]. Since the mechanisms of myofibroblasts which appear in damaged lung tissue is complex, we can not exclude the possibility that the decreased number of myofibroblasts in bleomycin-treated Smad3 knockout mice was either a result of the inhibition of these precursors of myofibroblast migration to the lung or the inhibition of the fibroblast-myofibroblast differentiation process.
Taken together, TGF-β1/Smad3 is a major pathway that regulates myofibroblast differentiation. This result indicates a potential significance for future attempts for attenuating the progression of human lung fibrosis by the inhibition of the Smad3 cascade.
References
- Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med 2001;345:517-25.
- King TE Jr, Schwarz MI, Brown K, Tooze JA, Colby TV, Waldron JA Jr, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001;164:1025-32.
- Phan SH. The myofibroblasts in pulmonary fibrosis. Chest 2001;120:S286-S9.
- Kuhn C, McDonald JA. The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol 1991;138:1257-65.
- Zhang K, Rekhter MD, Gordon D, Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am J Pathol 1994;145:114-25.
- Hashimoto S, Gon Y, Takeshita I, Matsumoto K, Maruoka S, Horie T. Tranaforming growth factor-β1 induces phenotypic modulation of human lung fibroblasts to myofibroblasts through a c-jun-NH2-terminal kinase-dependent pathway. Am J Respir Crit Care Med 2001;163:152-7.
- Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor beta1 induceds prolonged severe fibrosis in rat lung. J Clin Invest 1997;100:768-76.
- Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med 1994;331:1286-92.
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to nucleus. Cell 2003;113:685-700.
- Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science 2002;296:1646.
- Massague J. TGF-beta signal transduction. Annu Rev Biochem 1998;67:753-91.
- Dennler S, Huet S, Gauthier JM. A short amino-acid sequence in MH1 domain is responsible for functional differences between Smad2 and Smad3. Oncogene 1999;18:1643-8.
- Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, et al. Targeted disruption of SMAD3 results in impaired mucosol immunity and diminished T cell responsiveness to TGF-beta. EMBO J 1999;18:1280-91.
- Weinstein M, Yang X, Li C, Xu X, Gotay J, Deng CX. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc Natl Acad Sci USA 1998;95:9378-83.
- Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M, . Functional chrarcterization of transforming growth factorβ signaling in Smad2 and Smad3-deficient fibroblasts. J Biol Chem 2001; 276: 19 945–53.
- Liu X, Sun Y, Constantinescu SN, Karam E, Weinberg RA, Lodish HF. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc Natl Acad Sci USA 1997;94:10669-74.
- Zhao J, Shi W, Wang YL, Chen H, Bringas P Jr, Datto MB, et al. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 2002;282:L585-93.
- Stegemann H, Stalder K. Determination of hydroxyproline. Clin Chim Acta 1967;18:267-73.
- Ueno H, Sakamoto T, Nakamura T, Qi Z, Astuchi N, Takeshita A, et al. A soluble transforming growth factor beta receptor expressed in muscle prevents liver fibrogenesis in rats. Hum Gene Ther 2000;11:33-42.
- Giri SN, Hyde DM, Hollinger MA. Effect of transforming growth factor-β on a bleomycin induced accumulation of lung collagen in mice. Thorax 1993;48:959-66.
- Wang Q, Wang Y, Hyde DM, Gotwals PJ, Koteliansky VE, Ryan ST, et al. Reduction of bleomycin induced lung fibrosis by transforming growth factor-β soluble receptor in hamsters. Thorax 1999;54:805-12.
- Vindevoghel L, Lechleider RJ, Kon A, de Caestecker MP, Uitto J, Roberts AB, et al. SMAD3/4-dependent transcriptional activation of the human type VII collagen gene (COL7A1) promoter by transforming growth factor-β. Proc Natl Acad Sci USA 1998;95:14769-74.
- Chen SJ, Yuan W, Lo S, Trojanowska M, Varga J. Interaction of Smad3 with a proximal smad-binding element of the human alpha2(I) procollagen gene promoter required for transcriptional activation by TGF-beta. J Cell Physiol 2001;183:381-92.
- Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem 2001;276:17058-62.
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO 1998;17:3091-100.
- Datta PK, Blake MC, Moses HL. Regulation of plasminogen activator inhibitor-1 expression by transforming growth factor-beta-induced physical and functional interactions between smads and Sp1. J Biol Chem 2000;275:40014-9.
- Hu B, Wu Z, Phan SH. Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am J Respir Cell Mol Biol 2003;29:397-404.
- Corjay MH, Thompson MM, Lynch KR, Owens GK. Differential effect of platelet-derived growth factor-versus serum-induced growth on smooth muscle alpha-actin and nonmuscle beta-actin mRNA expression in cultured rat aortic smooth muscle cell. J Biol Chem 1998;264:10501-6.
- Mack CP, Owens GK. Regulation of smooth muscle alpha-actin expression in vivo is dependent on CArG elements within the 5' and first intron promoter regions. Circ Res 1999;84:852-61.
- Cogan JG, Subramanian SV, Polikandriotis JA, Kelm RJ, Strauch AR. Vascular smooth muscle α-actin gene transcription during myofibroblast differentiation requires sp1/3 protein binding proximal to the MCAT enhancer. J Biol Chem 2002; 277: 36 433–42.
- Yuan W, Varga J. Transforming growth factor-beta repression of matrix metal proteinase-1 transcription in dermal fibroblasts involves Smad3. J Biol Chem 2001;276:38502-10.
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signaling. Nature 2003;425:577-84.
- Sime PJ, O'Reilly KMA. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin immunol 2001;99:308-19.
- Liu T, Dhanasekaran SM, Jin H, Hu B, Tomlins SA, Chinnaiyan AM, et al. FIZZ1 stimulation of myofibroblast differentiation. Am J Path 2004;164:1315-26.
- Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol 2004;36:598-606.
- Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest 2004;113:243-52.