
From blood to brain: amoeboid microglial cell, a nascent macrophage and its functions in developing brain1
Introduction
Amoeboid microglial cells (AMC), the nascent form of ramified microglia that persist in the adult brain, are present ubiquitously in the brain during fetal and early postnatal development. They are present transiently until 10–14 d of age (Figure 1A) when all of them transform into the adult, ramified microglial cells (Figure 1B). There is recent evidence that the ramification of AMC may be regulated by cytoskeletal elements as transfection of the cells with a cytoskeleton-related gene, in particular, juxtanodin has resulted in the branching of the cells assuming a ramified external morphology (Figure 2). AMC exist singly or in clusters in the subventricular white matter. Other areas in which AMC are preponderant include the cavum septum pellucidum and subependymal cysts closely associated with the third and fourth ventricles and the cerebral aqueduct. In the brain tissue, the cells are widely distributed; some of them may be spatially associated with neurons, blood vessels, or dispersed freely. The close association of AMC to blood vessels has been reported in many areas of the brain[1–3].


The origin and mode of the formation of AMC has been a contentious issue for decades, and many theories had been proposed since the first description of a cellular “third element” other than neurons and neuroglia[4] and identification of microglia by del Rio-Hortega[4,5]. Three hypotheses have been put forward in relation to the origin of microglia: (i) mesodermal[6,7]; (ii) neuroectodermal[8–10]; and (iii) monocytic[11–13] . Investigations carried out in our laboratory over the past 3 decades tend to support the monocytic origin of AMC, although the possibility that these cells may be derived by direct invasion of fetal macrophages cannot be excluded[14]. We have shown that circulating monocytes invade the brain during embryonic and early postnatal life and then transform into AMC; hence, they are monocyte-derived brain macrophages like other tissue macrophages.
Nature of AMC
AMC are multifunctional immune cells in the developing central nervous system (CNS) that play an important role in the defense of neural parenchyma. Early studies have shown AMC to be active macrophages in the developing brain[13,15], removing cellular debris during normal development as well as in pathological conditions. Besides their scavenging function, AMC may also exert a cytotoxic effect through the secretion of toxic factors, such as nitric oxide[16]. Recent studies in our laboratory have greatly amplified the functional roles of these cells during development, in addition to their primary role in phagocytosis.
AMC function as phagocytes The phagocytic nature of AMC has been shown by various methods and observations, including the localization of hydrolytic enzymes, ultrastructural features shared by tissue macrophages, uptake of exogenous substances, and the activation of surface receptors and antigens related to phagocytosis.
Hydrolytic enzymes Our early cytochemical studies have shown the presence of hydrolytic enzymes, including acid phosphatase, aryl phosphatase, non-specific esterase and 5'-nucleotidase in the lysosomes in AMC[12,17,18]. The high contents of these hydrolytic enzymes in AMC suggested that these cells were active macrophages. This observation is supported by recent studies which have reported the localization of hydrolytic enzymes, such as acid phosphatase in macrophages, in the pineal gland[19]. A study investigating the ability of peritoneal, alveolar, and splenic macrophages and Kupffer cells to kill pathogens[20] reported that acid phosphatase activity was significantly increased in macrophages which ingested the pathogens and killed them. In vitro studies have shown that AMC were phagocytic and possessed non-specific esterase activity[21]. With time, these cells transform into ramified microglia-like cells and lose their phagocytic property as well as non-specific esterase content[21].
Ultrastructural observations Ultrastructural studies carried out in our laboratory in the normal postnatal brain and in the brains of rats subjected to hypoxia or Escheria coli(E coli) treatment have supported the phagocytic property of AMC. The macrophagic nature of AMC was evidenced by their engagement in the phagocytosis of degenerating axons and cells in the normal developing brain[22]. Under the electron microscope, some AMC in the corpus callosum of postnatal rats were observed to extrude portions of their cytoplasm which were phagocytosed by neighbouring AMC[22].
In the fetal and postnatal rat brains, AMC were found to phagocytose dead cells in the brain after transient maternal hypoxia[23]. Neonatal rats exposed to hypoxia also showed AMC in the corpus callosum engaged in the phagocytosis of apoptotic cells[24] and degenerating axons[16]. Further evidence of the phagocytic nature of AMC comes from their involvement in the removal of E coli introduced directly into the neonatal brain. Many of the injected E. coli were sequestered by AMC in less than 3 h[25]. It was concluded from these observations that AMC form a protective barrier which is deemed to be necessary in the early developmental period when the blood-brain barrier (BBB) is deficient.
Tracer studies It is well established that the BBB in the developing brain is immature. Administration of tracers, such as rhodamine isothiocyanate and horseradish peroxidase (HRP), intraperitoneally or intravenously, resulted in the labeling of AMC in the corpus callosum and other regions[26,27]. This suggested that substances in circulation leaked through the immature BBB and were phagocytosed by AMC. Another tracer, biotinylated dextran, when injected in various areas of the brain far removed from the corpus callosum, also resulted in the labeling of AMC[28]. It was concluded that AMC become labeled by ingesting the tracer which diffused slowly through the extracellular spaces from the injection site. Injection of HRP in the lumbosacral region of the spinal cord also resulted in the labeling of AMC in the corpus callosum[29]. The results suggested an ascending diffusion of the injected HRP in the spinal cord via wide interstitial spaces to reach the cerebrum where it was engulfed by the AMC.
ED1 antigens Further support to the macrophagic nature of AMC was lent by the expression of ED1 antigens on these cells. ED1 antigens are expressed by cells of monocyte/macrophage lineage[30]. These antigens were expressed by AMC in the fetal[31] and postnatal period[32], but not in adult rats.
Complement type 3 receptors Complement type 3 receptors (CR3) were detected on the cell membranes of AMC by using the antibody OX-42. It was proposed that these receptors are related to the active role of AMC in endocytosis[33]. This was supported by reports that CR3 receptors on monocytes and their derivative macrophages mediate endocytosis[34–37].
Lectin histochemistry Microglial cells engulfing pyknotic and fragmented nuclei of cells undergoing programmed cell death have also been visualized by use of lectin histochemical staining in conjunction with cresyl violet counterstaining[38].
Antigen presentation Although the CNS, under normal conditions, has been considered an immunologically privileged site for a long time, our studies showed for the first time that major histocompatibility class (MHC) I antigens were expressed by AMC[39]. MHC antigens are surface molecules required for the participation of macrophages in the activation of T lymphocytes by presenting certain antigens to them. MHC I antigens serve as restriction elements for cytotoxic/suppressor lymphocytes[40,41]. The expression of these antigens on AMC was related to their phagocytic activity[39]. The presence of MHC I antigens on AMC also suggests that these cells are ready to interact with infiltrating lymphocytes as the BBB is immature in the developing brain and the danger of a potential immune threat in early development may be present.
MHC II antigens, required for interaction with helper/inducer T lymphocytes, on the other hand, are not expressed by AMC under normal conditions. The expression of MHC II is induced under pathological and experimental conditions, for example, these antigens are expressed when the cells are challenged with lipopolysaccharide (LPS)[42] or with interferon-γ (IFN-γ)[43]. MHC II expression on AMC was also induced on the introduction of live E coli in their vicinity[25]. The expression of these antigens on AMC under pathological conditions suggests that they have the capability of interacting with helper/inducer cells to mount a potential immune response.
Other protective functions AMC in the developing brain express transferrin receptors[32] which facilitate the acquisition of iron needed for various functions of cells. As transferrin receptors are also important for the proliferation of cells, they may be involved in the differentiation and maturation of AMC. Additionally, the presence of these receptors on AMC may serve a protective function to sequester excess iron for storage in pathological conditions where there is an excessive influx of iron into the brain. In support of this, the increased expression of transferrin receptors and iron was reported[44] in AMC in newborn rats subjected to hypoxia. Hypoxia/reoxygenation is known to increase the iron content of the brain in newborn animals[45]. Excess iron not safely sequestered in storage is hazardous as it promotes the formation of free radicals[46], resulting in oxidative tissue damage.
AMC in the developing brain, especially in the white matter tracts, are also thought to promote axonal growth[47]. Cultured microglial cells derived from neonatal rat brains and transplanted into injured spinal cords enhanced the regeneration and elongation of spinal cord axons[48] indicating their involvement in axonal growth. In the developing white matter, AMC have also been reported to function as guides for developing axons, perhaps through the manufacture of extracellular matrix molecules, such as thrombospondin[49,50].
Insulin-like growth factors Insulin-like growth factor (IGF) I and II are known to regulate the development of the nervous system[51]. IGF-I plays an important role in promoting cell proliferation and differentiation[52] in the developing brain. The role of IGF II during postnatal development of the brain is less clear. Our recent study[53] has shown the expression of IGF-I and IGF-II in AMC. The expression of IGF-I and IGF-II was markedly enhanced in the cells by LPS, but was significantly suppressed with all-trans retinoic acid (RA). From these findings, it was suggested that IGF-I expression in AMC may be linked to the state of cell activation. IGF-I has been shown to enhance phagocytic activity of neutrophils in vitro when they were challenged with E coli[54]. It was demonstrated by Inoue et al[55] that IGF-I increased the killing capacity and phagocytosis of peritoneal macrophages when they were activated by E coli. IGF-I expression in AMC may also have paracrine functions, such as the modulation of the proliferation and development of the oligodendrocytes and myelination, as it has been considered as an important factor for oligodendrocyte survival and myelination[56,57]. Although the exact function of IGF-II in the developing brain is not clear, its pattern of association with oligodendrocytes and myelin suggests that it may also play a role in myelination[58,59] or in the phagocytic activity of AMC. IGF-I may also be related to the antigen-presenting function of AMC as it is known to stimulate the proliferation of immunocompetent cells and modulate the cellular immune functions of neutrophils and macrophages[60].
Nitric oxide Nitric oxide (NO) is synthesized from L-arginine by the family of NO synthase (NOS) enzymes. NOS from neurons and endothelial cells are constitutively expressed enzymes, the activities of which are calcium depend-ent. Inducible NOS (iNOS), which is calcium-independent and NO-generated from this isoform, is known to mediate immune functions. NO production in macrophages has been described to have protective or destructive functions. NO production by macrophages is stimulated by various pathogens, such as bacteria, viruses, and parasites[61–63] and has a role in their phagocytic activity[64]. The overproduction of NO however has toxic effects as it leads to the formation of toxic reactive nitrogen intermediates which can have deleterious effects[65].
It has been reported that activated microglial cells in the white matter may contribute to perinatal brain injury[66] through the secretion or production of noxious substances. NO production in the corpus callosum in response to hypoxia was found to increase in the neonatal brain along with iNOS expression in AMC[16,67]. In vitro studies have shown that NO produced by AMC is highly damaging to the oligodendrocytes resulting in their lysis[68]. The induction of iNOS in the activated microglia contributed to tissue injury through NO overproduction in fetal brain injury caused by umbilical cord occlusion[69]. The sustained production of NO endows macrophages with cytotoxic activity against viruses, bacteria, and fungi[70]. On the other hand, NO has suppressive effects on lymphocyte proliferation and can damage normal host cells[70] which can be deleterious.
Glutamate receptors The weak expression of NMDA (N-methyl-D-asparate) receptor subtype 1 (NMDAR1) was localized in AMC in the corpus callosum in the neonatal brain. This may facilitate the cells to be responsive to the release of glutamate from the neighboring callosal axons undergoing degeneration in the remodeling of the developing brain. The expression of NMDAR1 on AMC was enhanced in response to hypoxia[16]. This may be linked to the elevation of extracellular glutamate in the white matter in hypoxia/ischemia, which has been described to be possibly due to the release from damaged axons[71]. Excess glutamate leads to toxicity and death of oligodendrocyte progenitors[72]. Immunostimulated microglia have been reported to enhance the NMDA receptor-mediated excitotoxicity in part through the expression of iNOS[73]. The expression of NMDA receptors on microglial cells has also been reported in excitotoxic lesions in mice[74]. Although the expression of NMDAR1 receptors on AMC has been reported to have detrimental effects in the developing brain, they may have a protective role by sequestering excess glutamate released by degenerating axons.
Inflammatory response Microglial cells play an important role in the development of an inflammatory response in the developing brain[75]. They are thought to cause damage to axons and the developing oligodendrocytes by releasing inflammatory cytokines, such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α in many pathological conditions, such as hypoxic–ischemic conditions, and have been shown to express both p55TNFαR1 and p75TNFαR2 receptors[76]. Aberrant TNF-α/p55TNFαR1 signaling in the CNS can have a potentially major role in CNS pathologies in which oligodendrocyte death and demyelination is a primary pathological feature[77]. Besides their inflammatory actions, cytokines IL-1β and TNF-α may also be involved in the transcriptional activation of the iNOS gene[78,79]. Microglia in the developing brain have also been shown to express the chemokine receptor CCR5 until 2 weeks of age[80,81]. It was proposed that CCR5 were involved in microglial recruitment and activation during brain development and after neonatal brain injury, such as hypoxic-ischemic injury.
Endothelins Endothelins (ET), consisting of 3 subtypes, ET-1, ET-2, and ET-3, are multifunctional peptides produced by a wide variety of cells under normal and pathological conditions. Besides basal vasoconstriction, they are known to exert mitogenic and anti-apoptotic actions[82,83], as well as act as growth-promoting factors involved in embryonic and fetal development[84–86]. It has been reported that macrophages and monocytes act as a source of ET-1 production during infection and inflammation[87,88].
We have recently reported the expression of ET, especially ET-1 in AMC[89]. It was further demonstrated that the expression decreased in response to hypoxia. The stimulation of AMC with LPS enhanced the expression of ET-1. It was suggested that the expression of ET-1 may have auto-crine functions, such as synthesis and secretion of chemo-kines, including stromal derived factor-1α (SDF-1α) and monocyte chemoattractant protein-1 (MCP-1), or paracrine actions on the developing glial cells, neurons, and blood vessels bearing the ET receptors.
2',3'-cyclic nucleotide 3'-phosphodiesterase The expression of the 2',3'-cyclic nucleotide 3'-phosphodiesterase (CNPase), a myelin-associated enzyme commonly regarded as a selective marker for immature oligodendrocytes, was localized in AMC in the developing rat brain from prenatal d 18 to postnatal d 10[90]. Although the function of CNPase in AMC is not known, it may serve a cytoskeletal role to change the shape of the cells for migration or for the transportation of cytoplasmic materials. It may also be involved in the phagocytic function of AMC or in the secretion of pro-inflammatory cytokines and growth factors by them. CNPase may also be involved in transformation of the cells from the early round and amoeboid shape to a ramified form with growth.
Response to drugs
Glucocorticoids Glucocorticoids, which are potent anti-inflammatory and immunosuppressive drugs, have been reported to suppress the number and the phagocytic activity of macrophages[91]. The administration of glucorticoids, such as cortisone or dexamethasone, in postnatal rats resulted in a substantial reduction in the number of AMC in the corpus callosum[11,92]. The decrease in the number of AMC was attributed to the suppression of the number of circulating monocytes, as glucocorticoids are known to reduce their numbers[93]. Another effect of glucocorticoids on AMC was their premature differentiation or maturation into ramified microglia. However, the phagocytic activity or proliferation of AMC did not appear to be affected by glucocorticoid treatment[92]. Dexamethasone also inhibited microglial activation by downregulating the neurotoxic and pro-inflammatory mediators such as NO, TNF-α, and IL-6[94–96]. Preliminary results in our laboratory (unpublished data) demonstrated that dexamethasone inhibited the migration of microglial cells by suppressing the release of MCP-1, a chemokine which regulates the migration of activated microglial cells to the inflammatory sites in the CNS. It has been further shown that the downregulation of MCP-1 expression in activated microglial cells by dexamethasone was mediated via the MKP-1-dependent inhibition of the JNK and p38 mitogen-activated protein kinase pathways.
Chloroquine Chloroquine, an antimalarial drug with anti-inflammatory properties, has proven to be a beneficial therapeutic agent in certain inflammatory disorders[97]. Chloroquine is known to exert its anti-inflammatory effect by downregulating the synthesis of pro-inflammatory cytokines, such as TNF-α and IL-1β[98]. It also reduces the expression of MHC II on Kupffer cells[99]. Besides its anti-inflammatory actions, chloroquine inhibits lysosomal degradation and produces lysosomal changes resembling that of lysosomal storage disease in a variety of cells, including macrophages[100,101]. Increased vacuolation and the accumulation of lysosomes were observed in AMC in response to the intraperitoneal administration of chloroquine in 1-d-old rats. This was attributed to the failure of digestion of internalized substances. The phagocytic activity and antigen-presenting function of AMC, however, was enhanced in response to chloroquine as evidenced by the upregulation of CR3 receptors and MHC I antigens[102].
Colchicine The number of AMC in the corpus callosum of postnatal rats was reduced following colchicine treatment[103]. It also brought about an early differentiation of AMC into the ramified form. Colchicine is a microtubule-disrupting drug[104,105] and is known to induce apoptosis in different cell types. In vitro studies have shown that macrophages assume irregular profiles following depolymerization of microtubules by colchicine[106]. It has been hypothesized that microtubules are important for the secretion of lysosomal enzymes and the intracellular degradation of materials phagocytosed by macrophages[107]. It has also been suggested that microtubules are necessary for the fusion of lysosomes with endosomes which must precede intracellular digestion of phagocytosed materials[108], whereas others have reported that intact microtubules are not required for the lysosomal-endosomal fusion[109]. We found that the mitotic activity of AMC was suppressed by colchicine, but the phagocytic activity remained unaffected[103].
All-trans retinoic acid (RA) Recently, we have shown that the RA, a vitamin A metabolite, can suppress the LPS/β-amyloid-induced activation of microglial cells in primary culture by inhibiting the expression and production of TNF-α and NO[110]. It has been further shown that RA enhances the mRNA expression of TGF-β1, which acts as an immunosuppressor, and RA receptor (RAR) β1, and attenuates NF-κB translocation from the cytoplasm to the nucleus in activated microglial cells. It has been suggested that the inhibition of TNF-α and NO synthesis by RA in the activated microglia is mediated via the inhibition of NF-κB translocation, which could be caused by the upregulation of RAR and TGF-β1 gene expression. RA has also been shown to suppress the expression of IGF-I and IGF-II in activated microglia, indicating that RA is effective in inhibiting the action of a wide array of molecules specific for activated microglia[41]. In view of these results, it was suggested that RA could be considered a potential therapeutic agent that may inhibit the inflammatory response of microglia in neurodegenerative diseases.
Melatonin Melatonin, a neurohormone synthesized by the pineal gland, is known to have immunomodulatory actions[111,112]. In addition to its immunomodulatory actions, melatonin has also been reported to be important for phagocytosis under physiological conditions[113]. AMC showed a significant increase in cell numbers and upregulation of CR3, MHC I and MHC II, and CD4 antigens in response to melatonin administration[114], indicating enhanced endocytic and antigen-presenting capacity. The expression of these receptors and antigens returned to control levels on cessation of melatonin administration, suggesting that increased immune potentiality of the microglial cells and its maintenance requires the continuous action of the drug.
Toxic effects of AMC
It has been reported that microglial cells may be involved in causing apoptosis[47] in the developing brain. Although there is no direct evidence for this observation, microglial cells have been identified as the source of nerve growth factor, a pro-apoptotic agent responsible for neuronal death in the developing eye[115].
Conclusion
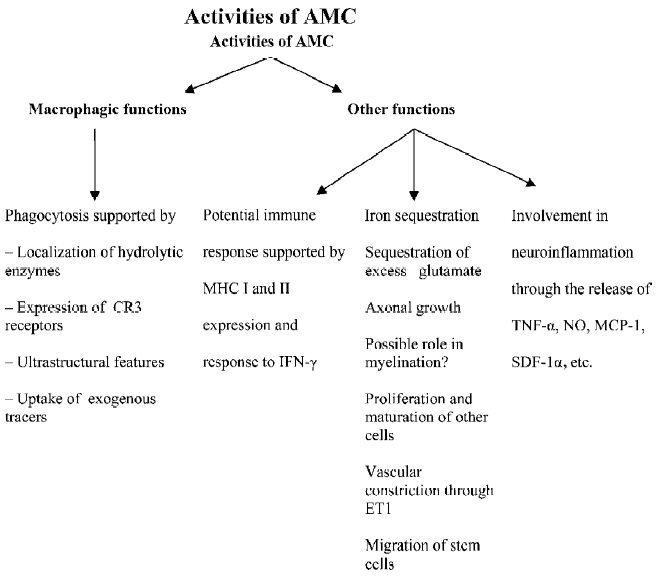
Early descriptions of the function of AMC focused on their primary role, that is, phagocytosis in the developing brain. In light of recent voluminous findings based on the expression of a plethora of molecules and growth factors in these cells, multiple functional roles of these cells, such as antigen presentation, vascular regulation, chemokine release, modulation of proliferation, and the development of other cells in the developing brain are proposed as summarized in Figure 3.
References
- Fujimoto E, Miki A, Mizoguti H. Histochemical study of the differentiation of microglial cells in the developing human cerebral hemispheres. J Anat 1989;166:253-64.
- Dalmau I, Finsen B, Zimmer J, Gonzalez B, Castellano B. Development of microglia in the postnatal rat hippocampus. Hippocampus 1998;8:458-74.
- Grossmann R, Stence N, Carr J, Fuller L, Waite M, Dailey ME. Juxtavascular microglia migrate along brain microvessels following activation during early postnatal development. Glia 2002;37:229-40.
- del Rio-Hortega P. Microglia. Cytology and cellular pathology of the nervous system; v 2 1932, pp 483-534. Ed. W. Penfield. PaulB. Hoeber, New York.
- del Rio-Hortega P. El “tercer elemento” de los centros nerviosus. Bol Soc Esp Biol 1919;9:69-120.
- Boya J, Calvo J, Prado A. The origin of microglial cells. J Anat 1979;129:177-86.
- Ashwell K. The distribution of microglia and cell death in the fetal rat forebrain. Brain Res Dev Brain Res 1991;58:1-12.
- Paterson JA., Privat A, Ling EA, Leblond CP. Investigation of glial cells in semithin sections. 3. Transformation of subependymal cells into glial cells, as shown by radioautography after 3H-thymidine injection into the lateral ventricle of the brain of young rats. J Comp Neurol 1973;149:83-102.
- Fujita S, Tsuchihashi Y, Kitamura T. Origin, morphology and function of the microglia. Prog Clin Biol Res 1981;59:141-69.
- Kitamura T, Miyake T, Fujita S. Genesis of resting microglia in the gray matter of mouse hippocampus. J Comp Neurol 1984;226:421-33.
- Ling EA. The origin and nature of microglia. In: Fedoroff S, Hertz L, editors. Advances in cellular neurobiology; v II. Xxxx: Academic Press; 1981. p 33–82.
- Ling EA, Kaur C, Wong WC. Light and electron microscopic demonstration of non-specific esterase in amoeboid microglial cells in the corpus callosum in postnatal rats: a cytochemical link to monocytes. J Anat 1982;135:385-94.
- Thomas WE. Brain macrophages: evaluation of microglia and their functions. Brain Res Brain Res Rev 1992;17:61-74.
- Kaur C, Hao AJ, Wu CH, Ling EA. Origin of microglia. Microsc Res Tech 2001;54:2-9.
- Ling EA, Wong WC. The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia 1993;7:9-18.
- Kaur C, Sivakumar V, Ang LS, Sundaresan A. Hypoxic damage to the periventricular white matter in neonatal brain: role of vascular endothelial growth factor, nitric oxide and excitotoxicity. J Neurochem 2006;98:1200-16.
- Ling EA. Light and electron microscopic demonstration of some lysosomal enzymes in the amoeboid microglia in neonatal rat brain. J Anat 1977;123:637-48.
- Kaur C, Ling EA, Wong WC. Cytochemical localisation of 5'-nucleotidase in amoeboid microglial cells in postnatal rats. J Anat 1984;139:1-7.
- Moller M, Rath MF, Klein DC. The perivascular phagocyte of the mouse pineal gland: an antigen-presenting cell. Chronobiol Int 2006;23:393-401.
- Black CM, Beaman BL, Donovan RM, Goldstein E. Intracellular acid phosphatase content and ability of different macrophage populations to kill Nocardia asteroides. Infect Immun 1985;47:375-83.
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci 1986;6:2163-78.
- Kaur C, Ling EA, Wong WC. Transformation of amoeboid microglial cells into microglia in the corpus callosum of the postnatal rat brain. An electron microscopical study. Arch Histol Jpn 1985;48:17-25.
- Li YB, Kaur C, Ling EA. Labeling of amoeboid microglial cells and intraventricular macrophages in fetal rats following a maternal injection of a fluorescent dye. Neurosci Res 1997;28:119-25.
- Kaur C, You Y. Ultrastructure and function of the amoeboid microglial cells in the periventricular white matter in postnatal rat brain following a hypoxic exposure. Neurosci Lett 2000;290:17-20.
- Kaur C, Too HF, Ling EA. Phagocytosis of Escherichia coli by amoeboid microglial cells in the developing brain. Acta Neuropathol (Berl) 2004;107:204-8.
- Kaur C, Ling EA, Wong WC. Labelling of amoeboid microglial cells in rats of various ages following an intravenous injection of horseradish peroxidase. Acta Anat (Basel) 1986;125:132-7.
- Xu J, Kaur C, Ling EA. Variation with age in the labelling of amoeboid microglial cells in rats following intraperitoneal or intravenous injection of a fluorescent dye. J Anat 1993;182:55-63.
- Earle KL, Mitrofanis J. Identification of transient microglial cell colonies in the forebrain white matter of developing rats. J Comp Neurol 1997;387:371-84.
- Leong SK, Shieh JY, Ling EA, Wong WC. Labelling of amoeboid microglial cells in the supraventricular corpus callosum following the application of horseradish peroxidase in the cerebrum and spinal cord in rats. J Anat 1983;136:367-77.
- Dijkstra CD, Dopp EA, Joling P, Kraal G. The heterogeneity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in rat recognized by monoclonal antibodies ED1, ED2 and ED3. Adv Exp Med Biol 1985;186:409-19.
- Wang CC, Wu CH, Shieh JY, Wen CY, Ling EA. Immunohistochemical study of amoeboid microglial cells in fetal rat brain. J Anat 1996;189:567-74.
- Kaur C, Ling EA. Transient expression of transferrin receptors and localisation of iron in amoeboid microglia in postnatal rats. J Anat 1995;186:165-73.
- Ling EA, Kaur C, Yick TY, Wong WC. Immunocytochemical localization of CR3 complement receptors with OX-42 in amoeboid microglia in postnatal rats. Anat Embryol (Berl) 1990;182:481-6.
- Newman SL, Musson RA, Henson PM. Development of functional complement receptors during in vitro maturation of human monocytes into macrophages. J Immunol 1980;125:2236-44.
- Beller DI, Springer TA, Schreiber RD. Anti-Mac-1 selectively inhibits the mouse and human type three complement receptor. J Exp Med 1982;156:1000-9.
- Abrahamson DR, Fearon DT. Endocytosis of the C3b receptor of complement within coated pits in human polymorphonuclear leukocytes and monocytes. Lab Invest 1983;48:162-8.
- Wright SD, Rao PE, van Voorhis WC, Craigmyle LS, Iida K, Talle MA, et al. Identification of the C3bi receptor of human monocytes and macrophages by using monoclonal antibodies. Proc Natl Acad Sci USA 1983;80:5699-703.
- Streit WJ. Role of macrophages and microglia in the injured CNS. In: Berry M, Logan A, editors. Pharmacological strategies in the treatment of CNS injuries. Boca Raton: CRC Press; 1999. p 81–97.
- Ling EA, Kaur C, Wong WC. Expression of major histocompatibility complex and leukocyte common antigens in amoeboid microglia in postnatal rats. J Anat 1991;177:117-26.
- Akiyama H, Itagaki S, McGeer PL. Major histocompatibility complex antigen expression on rat microglia following epidural kainic acid lesions. J Neurosci Res 1988;20:147-57.
- Weinstein DL, Walker DG, Akiyama H, McGeer PL. Herpes simplex virus type I infection of the CNS induces major histocompatibility complex antigen expression on rat microglia. J Neurosci Res 1990;26:55-65.
- Xu J, Ling EA. Upregulation and induction of surface antigens with special reference to MHC class II expression in microglia in postnatal rat brain following intravenous or intraperitoneal injections of lipopolysaccharide. J Anat 1994;184:285-96.
- Xu J, Ling EA. Upregulation and induction of major histocompatibility complex class I and II antigens on microglial cells in early postnatal rat brain following intraperitoneal injections of recombinant interferon-gamma. Neuroscience 1994;60:959-67.
- Kaur C, Ling EA. Increased expression of transferrin receptors and iron in amoeboid microglial cells in postnatal rats following an exposure to hypoxia. Neurosci Lett 1999;26:183-6.
- Adcock LM, Yamashita Y, Goddard-Finegold J, Smith CV. Cerebral hypoxia-ischemia increases microsomal iron in newborn piglets. Metab Brain Dis 1996;11:359-67.
- Gutteridge JM. Iron and oxygen: a biologically damaging mixture. Acta Paediatr Scand Suppl 1989;361:78-85.
- Streit WJ. Microglia and macrophages in the developing CNS. Neurotoxicology 2001;22:619-24.
- Rabchevsky AG, Streit WJ. Grafting of cultured microglial cells into the lesioned spinal cord of adult rats enhances neurite outgrowth. J Neurosci Res 1997;47:34-48.
- Chamak B, Dobbertin A, Mallat M. Immunohistochemical detection of thrombospondin in microglia in the developing rat brain. Neuroscience 1995;69:177-87.
- Chamak B, Morandi V, Mallat M. Brain macrophages stimulate neurite growth and regeneration by secreting thrombospondin. J Neurosci Res 1994;38:221-33.
- Fushimi S, Shirabe T. Expression of insulin-like growth factors in remyelination following ethidium bromide-induced demyelination in the mouse spinal cord. Neuropathology 2004;24:208-18.
- Guan J, Bennet L, Gluckman PD, Gunn AJ. Insulin-like growth factor-1 and post-ischemic brain injury. Prog Neurobiol 2003;70:443-62.
- Kaur C, Sivakumar V, Dheen ST, Ling EA. Insulin-like growth factor I and II expression and modulation in amoeboid microglial cells by lipopolysaccharide and retinoic acid. Neuroscience 2006;138:1233-44.
- Balteskard L, Unneberg K, Halvorsen D, Hansen JB, Revhaug A. Effects of insulin-like growth factor 1 on neutrophil and monocyte functions in normal and septic states. JPEN J Parenter Enteral Nutr 1998;22:127-35.
- Inoue T, Saito H, Fukushima R, Inaba T, Lin MT, Fukatsu K, et al. Growth hormone and insulin-like growth factor I enhance host defense in a murine sepsis model. Arch Surg 1995;130:1115-22.
- Dubois-Dalcq M, Murray K. Why are growth factors important in oligodendrocyte physiology? Pathol Biol (Paris) 2000;48:80-6.
- Guan J, Bennet L, George S, Wu D, Waldvogel HJ, Gluckman PD, et al. Insulin-like growth factor-1 reduces postischemic white matter injury in fetal sheep. J Cereb Blood Flow Metab 2001;21:493-502.
- Logan A, Gonzalez AM, Hill DJ, Berry M, Gregson NA, Baird A. Coordinated pattern of expression and localization of insulin-like growth factor-II (IGF-II) and IGF-binding protein-2 in the adult rat brain. Endocrinology 1994;135:2255-64.
- Walter HJ, Berry M, Hill DJ, Cwyfan-Hughes S, Holly JM, Logan A. Distinct sites of insulin-like growth factor (IGF)-II expression and localization in lesioned rat brain: possible roles of IGF binding proteins (IGFBPs) in the mediation of IGF-II activity. Endocrinology 1999;140:520-32.
- Auernhammer CJ, Strasburger CJ. Effects of growth hormone and insulin-like growth factor I on the immune system. Eur J Endocrinol 1995;133:635-45.
- Nathan C. Inducible nitric oxide synthase: what difference does it make? J Clin Invest 1997;100:2417-23.
- Green SJ, Mellouk S, Hoffman SL, Meltzer MS, Nacy CA. Cellular mechanisms of nonspecific immunity to intracellular infection: cytokine-induced synthesis of toxic nitrogen oxides from L-arginine by macrophages and hepatocytes. Immunol Lett 1990;25:15-9.
- Kremsner PG, Nussler A, Neifer S, Chaves MF, Bienzle U, Senaldi G, et al. Malaria antigen and cytokine-induced production of reactive nitrogen intermediates by murine macrophages: no relevance to the development of experimental cerebral malaria. Immunology 1993;78:286-90.
- Tumer C, Bilgin HM, Obay BD, Diken H, Atmaca M, Kelle M. Effect of nitric oxide on phagocytic activity of lipopolysaccharide-induced macrophages: possible role of exogenous l-arginine. Cell Biol Int in press.
- Eiserich JP, Patel RP, O’Donnell VB. Pathophysiology of nitric oxide and related species: free radical reactions and modification of biomolecules. Mol Aspects Med 1998;19:221-357.
- Leviton A, Gilles F. Ventriculomegaly, delayed myelination, white matter hypoplasia, and “periventricular” leukomalacia: how are they related? Pediatr Neurol 1996;15:127-36.
- You Y, Kaur C. Expression of induced nitric oxide synthase in amoeboid microglia in postnatal rats following an exposure to hypoxia. Neurosci Lett 2000;279:101-4.
- Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J Immunol 1993;151:2132-41.
- Ohyu J, Marumo G, Ozawa H, Takashima S, Nakajima K, Kohsaka S, et al. Early axonal and glial pathology in fetal sheep brains with leukomalacia induced by repeated umbilical cord occlusion. Brain Dev 1999;21:248-52.
- MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol 1997;15:323-50.
- Meng SZ, Arai Y, Deguchi K, Takashima S. Early detection of axonal and neuronal lesions in prenatal-onset periventricular leukomalacia. Brain Dev 1997;19:480-4.
- Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res 2001;50:553-62.
- Kim WK, Ko KH. Potentiation of N-methyl-D-aspartate-mediated neurotoxicity by immunostimulated murine microglia. J Neurosci Res 1998;54:17-26.
- Tahraoui SL, Marret S, Bodenant C, Leroux P, Dommergues MA, Evrard P, et al. Central role of microglia in neonatal excitotoxic lesions of the murine periventricular white matter. Brain Pathol 2001;11:56-71.
- Chew LJ, Takanohashi A, Bell M. Microglia and inflammation: impact on developmental brain injuries. Ment Retard Dev Disabil Res Rev 2006;12:105-12.
- Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol 1997;75:104-12.
- Akassoglou K, Douni E, Bauer J, Lassmann H, Kollias G, Probert L. Exclusive tumor necrosis factor (TNF) signaling by the p75TNF receptor triggers inflammatory ischemia in the CNS of transgenic mice. Proc Natl Acad Sci USA 2003;100:709-14.
- Lopez-Figueroa MO, Day HE, Lee S, Rivier C, Akil H, Watson SJ. Temporal and anatomical distribution of nitric oxide synthase mRNA expression and nitric oxide production during central nervous system inflammation. Brain Res 2000;852:239-46.
- Kadhim H, Khalifa M, Deltenre P, Casimir G, Sebire G. Molecular mechanisms of cell death in periventricular leukomalacia. Neurology 2006;67:293-9.
- Cowell RM, Xu H, Galasso JM, Silverstein FS. Hypoxic-ischemic injury induces macrophage inflammatory protein-1alpha expression in immature rat brain. Stroke 2002;33:795-801.
- Cowell RM, Xu H, Parent JM, Silverstein FS. Microglial expression of chemokine receptor CCR5 during rat forebrain development and after perinatal hypoxia-ischemia. J Neuroimmunol 2006;173:155-65.
- MacCumber MW, Ross CA, Snyder SH. Endothelin in brain: receptors, mitogenesis, and biosynthesis in glial cells. Proc Natl Acad Sci USA 1990;87:2359-63.
- Wu-Wong JR, Chiou WJ, Dickinson R, Opgenorth TJ. Endothelin attenuates apoptosis in human smooth muscle cells. Biochem J 1997;328:733-7.
- Yanagisawa M, Masaki T. Molecular biology and biochemistry of the endothelins. Trends Pharmacol Sci 1989;10:374-8.
- Ieda M, Fukuda K, Hisak Y, Kimura K, Kawaguchi H, Fujita J, et al. Endothelin-1 regulates cardiac sympathetic innervation in the rodent heart by controlling nerve growth factor expression. J Clin Invest 2004;113:876-84.
- Kedzierski RM, Yanagisawa M. Endothelin system: the double edged sword in health and disease. Annu Rev Pharmacol Toxicol 2001;41:851-76.
- Ebihara I, Nakamura T, Shimada N, Shoji H, Koide H. Effect of hemoperfusion with polymyxin B-immobilized fiber on plasma endothelin-1 and endothelin-1 mRNA in monocytes from patients with sepsis. Am J Kidney Dis 1998;32:953-61.
- Wahl JR, Goetsch NJ, Young HJ, van Maanen RJ, Johnson JD, Pea AS, et al. Murine macrophages produce endothelin-1 after microbial stimulation. Exp Biol Med 2005;230:652-8.
- Wu CY, Kaur C, Lu J, Cao Q, Guo CH, Zhou Y, et al. Transient expression of endothelins in the amoeboid microglial cells in the developing rat brain. Glia 2006;54:513-25.
- Wu CY, Lu J, Cao Q, Guo CH, Gao Q, Ling EA. Expression of 2',3'-cyclic nucleotide 3'-phosphodiesterase in the amoeboid microglial cells in the developing rat brain. Neuroscience 2006;142:333-41.
- Lortie C, King GM, Adamson IY. Effects of dexamethasone on macrophages in fetal and neonatal rat lung. Pediatr Pulmonol 1990;8:138-44.
- Kaur C, Wu CH, Wen CY, Ling EA. The effects of subcutaneous injections of glucocorticoids on amoeboid microglia in postnatal rats. Arch Histol Cytol 1994;57:449-59.
- Russo-Marie F. Macrophages and the glucocorticoids. J Neuroimmunol 1992;40:281-6.
- Chao CC, Hu S, Close K, Choi CS, Molitor TW, Novick WJ, et al. Cytokine release from microglia: differential inhibition by pentoxifylline and dexamethasone. J Infect Dis 1992;166:847-53.
- Golde S, Coles A, Lindquist JA, Compston A. Decreased iNOS synthesis mediates dexamethasone-induced protection of neurons from inflammatory injury in vitro. Eur J Neurosci 2003;18:2527-37.
- Lieb K, Engels S, Fiebich BL. Inhibition of LPS-induced iNOS and NO synthesis in primary rat microglial cells. Neurochem Int 2003;42:131-7.
- Potvin F, Petitclerc E, Marceau F, Poubelle PE. Mechanisms of action of antimalarials in inflammation: induction of apoptosis in human endothelial cells. J Immunol 1997;158:1872-9.
- Karres I, Kremer JP, Dietl I, Steckholzer U, Jochum M, Ertel W. Chloroquine inhibits proinflammatory cytokine release into human whole blood. Am J Physiol 1998;274:R1058-64.
- Ertel W, Morrison MH, Ayala A, Chaudry IH. Chloroquine attenuates hemorrhagic shock-induced suppression of Kupffer cell antigen presentation and major histocompatibility complex class II antigen expression through blockade of tumor necrosis factor and prostaglandin release. Blood 1991;78:1781-8.
- Mackenzie AH. Pharmacologic actions of 4-aminoquinoline compounds. Am J Med 1983;75:5-10.
- Korolenko TA, Rukavishnikova EV, Safina AF, Dushkin MI, Mynkina GI. Endocytosis by liver cells during suppression of intralysosomal proteolysis. Biol Chem Hoppe Seyler 1992;373:573-80.
- Kaur C, Wu CH, Singh J, Ling EA. Response of amoeboid microglial cells to chloroquine injections in postnatal rats. J Hirnforsch 1996;37:233-42.
- Kaur C. Effects of colchicine on amoeboid microglial cells in the postnatal rat brain. Arch Histol Cytol 1997;60:453-62.
- Tsukidate K, Yamamoto K, Snyder JW, Farber JL. Microtubule antagonists activate programmed cell death (apoptosis) in cultured rat hepatocytes. Am J Pathol 1993;143:918-25.
- Bonfoco E, Ceccatelli S, Manzo L, Nicotera P. Colchicine induces apoptosis in cerebellar granule cells. Exp Cell Res 1995;218:189-200.
- Rosania GR, Swanson JA. Microtubules can modulate pseudopod activity from a distance inside macrophages. Cell Motil Cytoskeleton 1996;34:230-45.
- Cohn ZA, Benson B. The in vitro differentiation of mono-nuclearphagocytes. I. The influence of inhibitors and the results of autoradiography. J Exp Med 1965;121:279.
- Malawista SE, Bodel PT. The dissociation by colchicine of phagocytosis from increased oxygen consumption in human leukocytes. J Clin Invest 1967;46:786.
- Pesanti EL, Axline SG. Colchicine effects on lysosomal enzyme induction and intracellular degradation in the cultivated macrophage. J Exp Med 1975;141:1030-46.
- Dheen ST, Jun Y, Yan Z, Tay SS, Ling EA. Retinoic acid inhibits expression of TNF-alpha and iNOS in activated rat microglia. Glia 2005;50:21-31.
- Maestroni GJ. The immunotherapeutic potential of melatonin. Expert Opin Investig Drugs 2001;10:467-76.
- Pandi-Perumal SR, Srinivasan V, Maestroni GJ, Cardinali DP, Poeggeler B, Hardeland R. Melatonin: nature’s most versatile biological signal? FEBS J 2006;273:2813-38.
- Hriscu ML. Modulatory factors of circadian phagocytic activity. Ann N Y Acad Sci 2005;1057:403-30.
- Kaur C, Ling EA. Effects of melatonin on macrophages/microglia in postnatal rat brain. J Pineal Res 1999;26:158-68.
- Frade JM, Barde YA. Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron 1998;20:35-41.