
Requirement of reactive oxygen species generation in apoptosis of leukemia cells induced by 2-methoxyestradiol
Introduction
Acute myelogenous leukemia is a serious and often lethal disease. There is a need for new, effective, and relatively non-toxic therapeutic modalities. Recent research has focused on targeted the disruption of proliferation signal transduction and the utility of 2-methoxyestradiol (2-ME) in the treatment of leukemia. 2-ME is a physiological metabolite of the endogenous estrogen estradiol-17β which fails to bind the estrogen receptor[1]. This compound is currently used in clinical trials as a single agent or in combination with other anticancer agents, for treatment of several types of human cancer, including breast cancer, prostate cancer, and multiple myeloma[2,3]. 2-ME also shows antileukemic activity in vitro with therapeutic selectivity[4].
2-ME is another inhibitor of microtubule dynamic causing mitotic arrest followed by cell cycle G2/M-phase arrest[5]. It was also found to have an anti-angiogenesis effect in tumors through its direct cytotoxicity on endothelial cells[6,7]. It has also been reported that 2-ME induces apoptosis in various cell types. In addition, recent studies have shown that 2-ME inhibits the transcription of the superoxide dismutase (SOD) enzymes, which protect cells from damage induced by superoxide radicals, and the inhibition of SOD activity results in apoptosis of human leukemia cells[8,9]. Because 2-ME is relatively non-toxic to normal tissues[9], an important implication of these findings is that such agents might play a useful role in the therapy of leukemia.
Two major apoptotic pathways are known to date. The intrinsic pathway is initiated by the mitochondria, while the extrinsic pathway is initiated by cell surface receptors[10]. Mitochondria-mediated apoptosis occurs in response to a wide range of stimuli. Mitochondria are the major generators of ATP by oxidative phosphorylation. Reactive oxygen species (ROS) are generated in and around mitochondria, and they are buffered by antioxidants. ROS may not only arise from endogenous sources, but also from exogenous sources. Our previous studies suggested that manumycin increases ROS levels and results in apoptosis[11]. Although many studies have shown that 2-ME has an effect in various tumor cells, including leukemia cells, currently the role of mitochondrial functional changes in the response of leukemia cells to 2-ME has not yet been explored.
We hypothesized that 2-ME would induce functional changes in the mitochondria in association with ROS generation in the course of apoptosis induction in leukemia cells. To test this hypothesis, we evaluated the effect of 2-ME on mitochondrial membrane potential, ROS, and apoptosis in leukemia cell lines. Our results indicated the requirement of ROS generation in apoptosis induced by 2-ME in leukemia cells.
Materials and methods
Cell culture Human leukemia cell lines HL-60 and U937 from American Type Culture Collection (Manassas, VA, USA) were maintained in suspension culture with RPMI-1640 medium supplemented with heat-treated fetal bovine serum (10%) at 37 oC in a humidified atmosphere with 5% CO2. All experiments were done in exponentially growing cultures.
Chemicals and reagents Manumycin, N-acetyl-L-cysteine (NAC), 2-ME, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium (MTT), carbonyl cyanide 4-phenylhydrazone (FCCP), and DMSO were purchased from Sigma Chemical Co (St Louis, MO, USA). All tissue culture media were purchased from Life Technologies (Gaithersburg, MD, USA). ApoAlert Nitric Oxide/Annexin V Dual Sensor Kits were purchased from BD Biosciences Clontech Laboratories (Palo Alto, CA, USA). 5,5',6,6'-Tetrachloro-1,1',3,3'-tetraethylben-zimidazolylcarbocyanine iodide (JC-1)and dihydroethidium (DHE) were purchased from Molecular Probes (Eugene, OR, USA) and prepared separately in DMSO. The stock solutions were then further diluted in tissue culture medium to the desired concentrations. 2-ME and manumycin were first dissolved in DMSO (tissue-culture grade). The stock solution was diluted in tissue culture medium at appropriate concentrations such that the final concentration of DMSO in culture medium would not exceed 0.1% (v/v).
Assessment of mitochondrial membrane potential Measurement of mitochondrial membrane potential (Dym) was performed with the JC-1 stain, a lipophilic cation fluorescent dye that accumulates in the mitochondria, showing green fluorescence at a low mitochondrial membrane potential and forming red fluorescent J-aggregates at higher membrane potential. Drop in mitochondrial membrane potential is indicated by a decrease in the ratio of the red signal to the green signal. HL-60 and U937 cells were cultured in 96-well tissue culture plates with black walls and clear bottoms. After attached to the vessels overnight, the cells were treated with experimental treatments, 10 µmol/L FCCP (positive control for depolarization), or 0.1% DMSO (negative control) for various durations. The cells were then incubated with 5 μg/mL JC-1 at 37 °C for 15 min in dark as previously described. The cells were washed twice with phosphate buffered solution (PBS), and the plates were immediate read using a fluorescent plate reader (Spectrafluor Plus, Tecan, Zurich, Switzer-land) with the excitation and emission wavelengths set at 540 and 595 nm, respectively for red fluorescence, and 485 and 535 nm, respectively for green fluorescence. For each sample, the results were calculated as follows after the fluorescence values had been corrected for the background:red fluorescence of sample-average of red fluorescence of blanks)/(green fluorescence of sample-green fluorescence-average of green fluorescence of blanks. Data from 3 independent experiments were analyzed using one-way ANOVA with a post-hoc comparison between the groups.
Flow cytometry assay Flow cytometry assays were performed at our Flow Cytometry Core Facility (Becton Dickinson, Mountain View, CA, USA). Phycoerythrin (PE)-conjugated Annexin V was used to detect cells undergoing apoptosis. In brief, the cells were collected by centrifugation (200×g for 5 min at 4 oC) after the experimental treatments and resuspended in Annexin V binding buffer with Annexin V-PE staining for 15 min at room temperature. After dilution, the cells were analyzed with flow cytometry for PE fluorescence (excitation wavelength, 488 nm; emission wavelength, 578 nm). To measure changes of intracellular nitric oxide (NO) level, a proprietary membrane-permeable NO sensor dye was used according to the manufacturer’s instructions. Briefly, the cells were pre-incubated with the NO sensor dye for 30 min prior to experimental treatments in the continued presence of the dye. The cells were pelleted by centrifugation, rinsed, and analyzed with flow cytometry for green fluorescence (excitation wavelength, 488 nm; emission wavelength, 515 nm). For dual NO/Annexin V simultaneous flow cytometry analysis, the ApoAlert Nitric Oxide/Annexin V Dual Sensor Kit was used.
Measurement of intracellular superoxide anion The cells were cultured in 96-well plates. HL-60 and U937 cells were treated with 2 µmol/L 2-ME for various durations, and then the cells were incubated with 5 μmol/L DHE as previously described. In brief, after experimental treatments, the cells were incubated with 5 µmol/L DHE in culture medium for 10 min at 37 °C in dark. The cells were rinsed twice with PBS, and then ethidium-DNA fluorescence was immediately measured with a fluorescent plate reader (Spectrafluor Plus, Tecan, Switzerland) with an excitation wavelength at 430 nm and an emission wavelength at 590 nm. The average ethidium-DNA fluorescence of each group with the 95% confidence intervals was plotted over time. Three independent experiments were performed
Cytotoxicity assays Cell growth inhibition or the relative number of viable cells was determined by MTT assay as previously described. In brief, HL-60 or U937 cells were seeded onto 96-well plates at the initial density of 5000 cells/well. After incubation with various concentrations of 2-ME, NAC, and a combination of both, respectively for 48 h, 20 µL MTT reagent (3 mg/mL) was added to each well and incubated at 37 oC for 4 h. The cells were then centrifuged (700×g for 15 min), and the medium was removed. The cell pellets were dissolved in 200 µL DMSO. Absorbance was measured at a wavelength of 590 nm with reference at wavelength of 635 nm. All of the experiments were performed with the absorbance values within the linear range of this colorimetric assay. Percentage viability was defined as 100% times the ratio of absorbance above the background in the sample to the average absorbance above the background in the control (DMSO-treated) samples. The background absorbance was measured in the wells with the cells that had been killed by exposure to 70% ethanol for 10 min.
Statistical analysis Comparison in experiments was performed using one-way ANOVA with Tukey’s test or Student’s t-test to assess the statistical significance of differences between the groups. Differences with P<0.05 were considered significant.
Results
2-ME decreased viability and induced apoptosis of leukemia cells 2-ME decreased the viability in the leukemia cell lines (Figure 1A). Modest degrees of cytotoxicity were noted at 1 µmol/L 2-ME (48 h), which reached near maximal levels of cytotoxicity at concentrations more than 2 µmol/L. Meanwhile, the time course of 2-ME-mediated apoptosis in HL-60 and U937 cells was analyzed (Figure 2B). Apoptosis-induced by 2 µmol/L 2-ME treatment became apparent after 12 h, and apoptosis was enhanced while the drug exposure time was elongated.
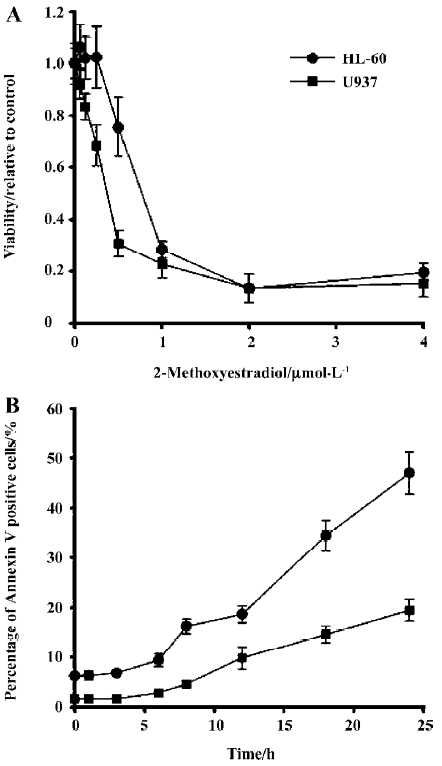
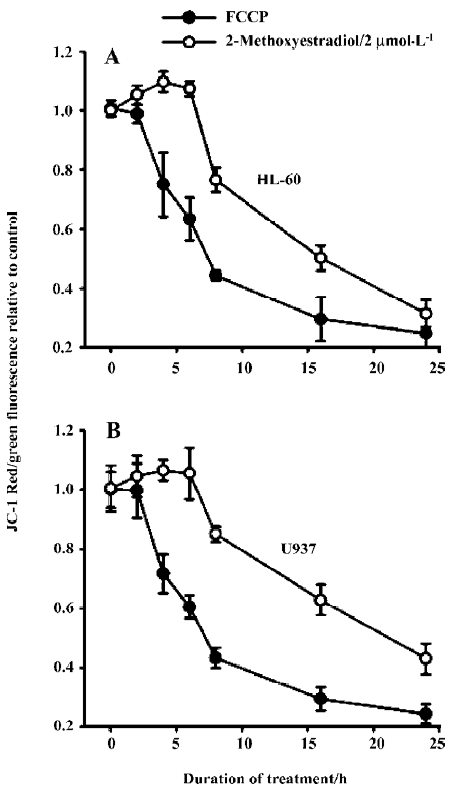
Effects of 2-ME on mitochondrial membrane potential It is well established that apoptosis induced by some agents is associated with the perturbation of mitochondrial functions. Therefore, we investigated changes in mitochondrial function in response to 2-ME treatment. Using the fluorescent dye JC-1, we assessed relative changes in mitochondrial membrane potential following drug treatments in leukemia cell lines. In both HL-60 and U937 cells, treatment with 2 µmol/L 2-ME induced mitochondrial membrane hyperpolarization in the early phase of treatment followed by depolari-zation. The positive control for depolarization was achieved by treating the cells with FCCP (Figure 2).
2-ME induced ROS generation in leukemia cells
2-ME induced NO generation in HL-60 and U937 cells 2-ME 2 µmol/L induced NO generation as demonstrated by the significant increase (P<0.05, ANOVA, post-hoc Tukey’s test) in the percentage of cells stained by NO sensor dye in 2-ME-treated cells compared with the DMSO sham-treated control cells (Figure 3A)
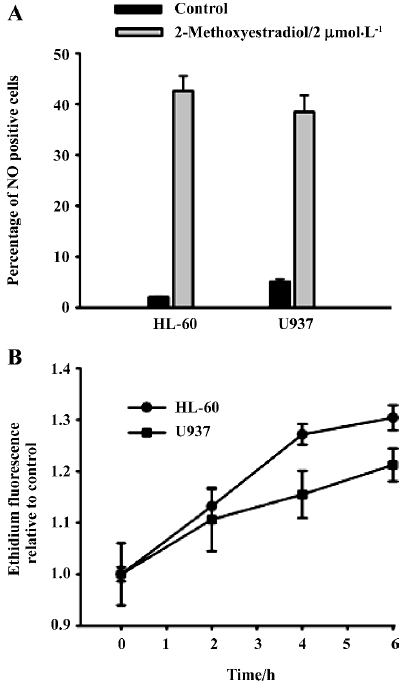
2-ME induced generation of superoxide anion Another major ROS is the superoxide anion. To determine whether 2-ME increased superoxide anion in leukemia cell lines, we measured superoxide anion by detecting the oxidation of DHE. HL-60 and U937 cells were exposed to 2 µmol/L 2-ME for 2, 4, or 6 h. Three independent experiments were performed for each cell line. 2-ME significantly increased superoxide anion levels relative to levels in untreated control cells at all 3 time points (Figure 3B).
2-ME induced apoptosis was associated with the generation of ROS in leukemia cells 2-ME induced NO generation before the cells underwent apoptosis. A dual-fluorescence flow cytometry analysis was used to determine the relationship between NO and apoptosis. The cells were incubated with 2 µmol/L 2-ME for various times and then double-stained with NO sensor dye and Annexin V. A substantial amount of NO accumulation was observed after 1 h of 2-ME treatment, as evidenced by a shift of the cell population from the lower-left quadrant, rightward along the x-axis. However, no detectable drug-induced apoptosis was observed at the same time (1 h), as evidenced by the lack of change in Annexin V labeling. In the continuous presence of 2-ME, NO remained elevated up to 6 h, whereas a significant number of Annexin V-positive cells did not appear until 6 h, and cell apoptosis was further increased at 6 h (Figure 4A). Similar results were obtained with U937 cells (data not shown). These results suggested that treatment with 2-ME in both cell lines would induce NO generation before the cells underwent apoptosis.
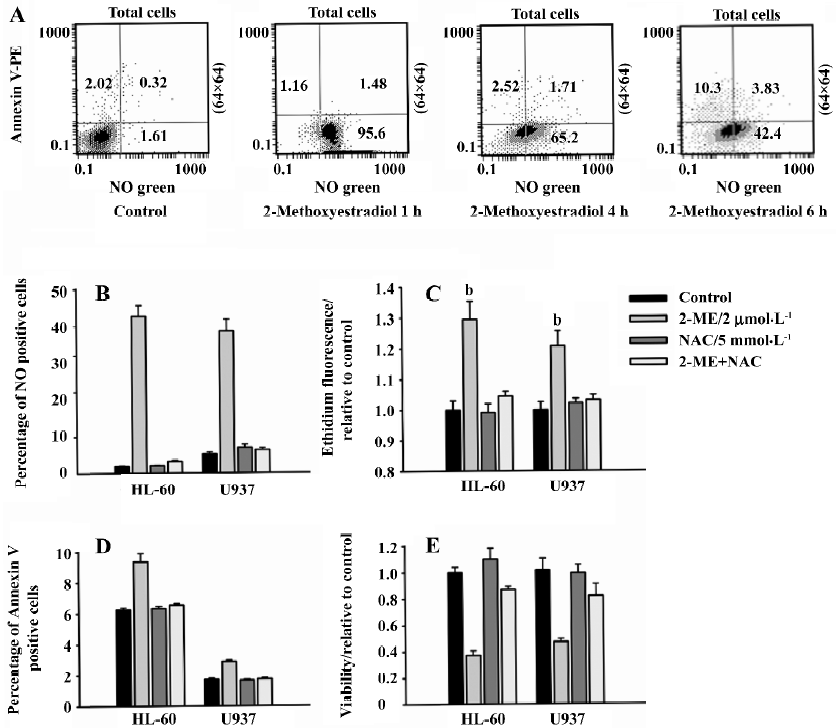
NAC blocked 2-ME-induced ROS generation NAC is a commonly used quencher of ROS. The ability of NAC to block increases in superoxide anion and NO has previously been documented. Co-culture with 5 mmol/L NAC for 6 h effectively blocked 2-ME-induced NO and superoxide anion generation in HL-60 and U937 cells (Figure 4B, 4C). These findings indicate that we could use NAC to demonstrate whether ROS is essential for 2-ME-induced apoptosis.
NAC blocked 2-ME-induced apoptosis HL-60 and U937 cells were incubated with 2 µmol/L 2-ME for 6 h, and apoptosis was detected by Annexin V-PE. 2-ME significantly (P<0.05, ANOVA, post-hoc Tukey’s test) increased the percentage of cells stained by Annexin V-PE compared with binding in the corresponding DMSO sham-treated control. At 5 mmol/L, NAC did not have effect on apoptosis, but the presence of NAC almost completely suppressed 2-ME-induced apoptosis, as evidenced by a near-complete reversal of the percentage of Annexin V-positive cells (Figure 4D). Taken together, these findings suggest that an increase in ROS is required for 2-ME-induced apoptosis in leukemia cells.
NAC protects leukemia cells from 2-ME cytotoxicity To further investigate whether NAC could protect leukemia cells from 2-ME cytotoxicity, we measured the relative viability of cells treated for 48 h with control medium: 2 µmol/L 2-ME, 5 mmol/L NAC, or a combination of both, respectively. The results of 3 independent experiments were plotted. Concurrent treatment with NAC significantly prevented the decrease in viability with 2-ME treatment (P<0.05; Figure 4E). These results suggest that the effect of 2-ME on viability was mostly mediated by ROS.
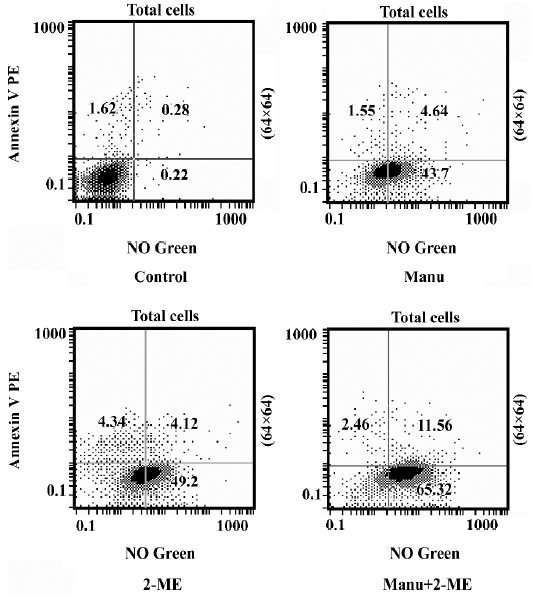
Manumycin enhanced 2-ME-induced NO generation and apoptosis We previously found that manumycin induced apoptosis of leukemia cells via the generation of NO. We also found 2-ME induced apoptosis of leukemia cells through the functional change of mitochondria and the increase in NO and superoxide anion. Therefore, we combined manumy-cin with 2-ME to evaluate the enhancement of apoptosis. Manumycin significantly increased the percentage of cells stained with the NO sensor dye only and the percentage of cells stained positive by both Annexin V-PE and the NO sensor dye in HL-60 cells. Compared with 2-ME alone, the combination of manumycin and 2-ME further increased the percentage of cells stained with the NO sensor dye only and the percentage of cells stained by both Annexin V-PE and the NO sensor dye (one-way ANOVA, post-hoc between-group comparisons, Tukey’s test, P<0.05; Figure 5). A similar result was obtained for the U937 cells (data not shown).
Discussion
We demonstrated here that 2-ME induced apoptosis in leukemia cells through the generation of ROS. This interpretation was based on the increase in the 2-ME-treated leukemia cells and on the significant protection against apoptosis exerted by NAC, a quencher of ROS. Our results corroborated and extended the findings of Huang et al on the induction of oxygen free radicals, mitochondria damage, and apoptosis in chronic lymphocytic leukemia[4–9].
It is clear that changes of mitochondrial membrane potential are associated with apoptosis[12–14]. Both hyperpolarization and depolarization have previously been observed in association with mitochondrial cytochrome c release[15]. However, mitochondrial depolarization in association with apoptosis appears to be more common. Resveratrol induces apoptosis with mitochondrial depolarization in acute lymphocytic leukemia cells[16]. Oxidative damage and mitochondrial depolarization induced by imexon highly correlates with imexon-induced apoptosis in several myeloma cell lines and an acute promyelocytic leukemia cell line[17]. The mitochondrial permeability transition (MPT) pore opening is expected to affect mitochondrial membrane potential. Vieira et al proposed a 2-step model for MPT in which hyperpolarization was associated with step 1 and non-specific pore opening and depolarization was associated with step 2. We found that 2-ME induced hyperpolarization at an early stage and subsequent depolarization in Hl-60 and U937 cells. It is conceivable that the duration of step 1 is very brief in HL-60 and U937 cells. The increase in superoxide also appears to correlate with mitochondrial membrane hyperpolarization induced by 2-ME.
Mitochondria are the rich source of ROS, which are toxic byproducts of aerobic cells and play an important role in cell proliferation, aging, and cancer development. An excessive amount of ROS can lead to cell death by apoptosis or by necrosis. The ability of ROS to damage cellular components and cause cell death suggests the possibility to explore this chemical for killing cancer cells[18]. The importance of chemotherapy-induced change in redox status to signal apoptosis and regulate the apoptosis effector is emerging. Although the role of ROS in apoptosis induction has been described for a diverse collection of xenobiotics, this report provides experimental evidence for the role of ROS in apoptosis induced by 2-ME. The mechanisms of action of 2-ME may involve the inhibition of Akt and the activation of Jun N-terminal kinase (JNK), the inhibition of SOD, anti-angiogenetic effects, and interruption microtubule assembl-ing. However, the question as to which one induces ROS generation is unclear. Future work to bridge this gap in knowledge will be important because this knowledge will provide the theoretical basis for the rational design of synergistic combination chemotherapy regimens, including 2-ME.
Superoxide and NO are the major ROS in living cells, and superoxide is the first species to be generated by the auto-oxidation of the mitochondria redox groups. ROS, including NO and superoxide anion generated by 2-ME, serves as stimuli to induce apoptosis in leukemia cells. NO in particular can be both pro- and anti-apoptotic depending on circumstances. We studied the production of NO and superoxide anion in HL-60 and U937 cells treated with 2-ME. Our results provided evidence of apoptosis induced by 2-ME. NAC blocked apoptosis induced by 2-ME indicated that ROS was a requirement in the apoptotic signaling cascade.
Farnesyltransferase inhibitors (FTIs) are a group of new chemotherapeutic agents being studied in clinical trials for both solid tumors and hematological malignancies. Manumycin is an FTI that mimics the farnesyl pyrophosphate group[21]. FTI have demonstrated preclinical activity in a variety of experimental models, and we previously demonstrated that manumycin induced ROS generation, which mediated DNA damage and induced apoptosis of anaplastic thyroid cancer (ATC) cells and leukemia cell lines. Oxidative damage has been suggested to be the key mechanism by which manumycin induced apoptosis. On the basis of the ability of manumycin to induce the generation of ROS in several experiment models, it is reasonable to expect that its combination with 2-ME could enhance apoptosis. The results of the present study demonstrated that the combination of manumycin with 2-ME enhanced the generation of NO and resulted in enhanced apoptosis induction in leukemia cells. This result from the in vitro assay suggested that it was possible to combine 2-ME with other ROS-generating agents to enhance the therapeutic effect on leukemia cells.
In conclusion, 2-ME induced oxidative stress and mitochondrial damage, which then caused apoptosis in leukemia cells. The mechanism of apoptosis induction by 2-ME involved the generation of ROS. The ROS-generating agent manumycin significantly enhanced apoptosis induced by 2-ME. These results may help to elucidate the mechanisms underlying efficacious combinations of 2-ME with other drugs and provide a new strategy to enhance therapeutic activity. Future studies will further investigate this possibility.
References
- LaVallee TM, Zhan XH, Herbstritt CJ, Kough EC, Green SJ, Pribluda VS. 2-Methoxyestradiol inhibits proliferation and induces apoptosis independently of estrogen receptors alpha and beta. Cancer Res 2002;62:3691-7.
- Ray G, Dhar G, van Veldhuizen PJ, Banerjee S, Saxena NK, Sengupta K, et al. Modulation of cell-cycle regulatory signaling network by 2-methoxyestradiol in prostate cancer cells is mediated through multiple signal transduction pathways. Biochemistry 2006;45:3703-13.
- Dahut WL, Lakhani NJ, Gulley JL, Arlen PM, Kohn EC, Kotz H, et al. Phase I clinical trial of oral 2-methoxyestradiol, an anti-angiogenic and apoptotic agent, in patients with solid tumors. Cancer Biol Ther 2006;5:22-7.
- Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003;101:4098-104.
- Lin HL, Liu TY, Chau GY, Lui WY, Chi CW. Comparison of 2-methoxyestradiol-induced, docetaxel-induced, and paclitaxel-induced apoptosis in hepatoma cells and its correlation with reactive oxygen species. Cancer 2000;89:983-94.
- Fotsis T, Zhang Y, Pepper MS, Adlercreutz H, Montesano R, Nawroth PP, et al. The endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature 1994;368:237-9.
- Robinson MR, Baffi J, Yuan P, Sung C, Byrnes G, Cox TA, et al. Safety and pharmacokinetics of intravitreal 2-methoxyestradiol implants in normal rabbit and pharmacodynamics in a rat model of choroidal neovascularization. Exp Eye Res 2002;74:309-17.
- Pani G, Colavitti R, Bedogni B, Fusco S, Ferraro D, Borrello S, et al. Mitochondrial superoxide dismutase: a promising target for new anticancer therapies. Curr Med Chem 2004;11:1299-308.
- Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 2000;407:390-5.
- Piwocka K, Vejda S, Cotter TG. GC OS, McKenna SL. Bcr-Abl reduces endoplasmic reticulum (ER) releasable calcium levels by a Bcl-2 independent mechanism and inhibits calcium dependent apoptotic signaling. Blood 2006;107:4003-10.
- She M, Pan I, Sun L, Yeung SC. Enhancement of manumycin A-induced apoptosis by methoxyamine in myeloid leukemia cells. Leukemia 2005;19:595-602.
- Scarlett JL, Sheard PW, Hughes G, Ledgerwood EC, Ku HH, Murphy MP. Changes in mitochondrial membrane potential during staurosporine-induced apoptosis in Jurkat cells. FEBS Lett 2000;475:267-72.
- Budd SL, Castilho RF, Nicholls DG. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett 1997;415:21-4.
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta 1995;1241:139-76.
- Poppe M, Reimertz C, Dussmann H, Krohn AJ, Luetjens CM, Bockelmann D, et al. Dissipation of potassium and proton gradients inhibits mitochondrial hyperpolarization and cytochrome c release during neural apoptosis. J Neurosci 2001;21:4551-63.
- Dorrie J, Gerauer H, Wachter Y, Zunino SJ. Resveratrol induces extensive apoptosis by depolarizing mitochondrial membranes and activating caspase-9 in acute lymphoblastic leukemia cells. Cancer Res 2001;61:4731-9.
- Dvorakova K, Waltmire CN, Payne CM, Tome ME, Briehl MM, Dorr RT. Induction of mitochondrial changes in myeloma cells by imexon. Blood 2001;97:3544-51.
- Pan J, She M, Xu ZX, Sun L, Yeung SC. Farnesyltransferase inhibitors induce DNA damage via reactive oxygen species in human cancer cells. Cancer Res 2005;65:3671-81.
- She M, Yang H, Sun L, Yeung SC. Redox control of manumycin A-induced apoptosis in anaplastic thyroid cancer cells: involvement of the xenobiotic apoptotic pathway. Cancer Biol Ther 2006;5:275-80.
- She M, Jim Yeung SC. Combining a matrix metalloproteinase inhibitor, a farnesyltransferase inhibitor, and a taxane improves survival in an anaplastic thyroid cancer model. Cancer Lett 2006;238:197-201.
- Kouchi H, Nakamura K, Fushimi K, Sakaguchi M, Miyazaki M, Ohe T, et al. Manumycin A, inhibitor of ras farnesyltransferase, inhibits proliferation and migration of rat vascular smooth muscle cells. Biochem Biophys Res Commun 1999;264:915-20.