
Molecular mechanisms of polypeptide from Chlamys farreri protecting HaCaT cells from apoptosis induced by UVA plus UVB1
Introduction
Increasing evidence over the past decade has indicated that UV irradiation provokes premature aging, cystitis, and skin cancer, which reinforces the need for novel chemoprevention strategies involving the use of anti-oxidants or other biological events occurring following UV exposure to the skin. UV that reaches the earth’s surface consists of 90%–99% UVA (wavelength 320–400 nm), 1%–10% UVB (290–320 nm), and a very small portion of UVC[1]. UVB penetrates the epidermis, and to a lesser extent, the upper part of the dermis, while UVA penetrates more deeply into human skin. Although UVC can damage skin to a larger extent than UVA and UVB, most of it is absorbed by ozone. In the present study, we irradiated cells by UVA plus UVB to mimic the UV light in the sunlight.
Relative molecular mass of polypeptide from Chlamys farreri (PCF) is 912. PCF is a novel, marine-active product isolated from the Chinese scallop Chlamys farreri, which has been served as a seafood for thousands of years. The extraction yield of PCF was 1.14 g/kg Chlamys farreri by our techniques. As an octapeptide, PCF consists of Pro, Asn, Ser, Thr, Arg, Lys, and Cys. However, the primary structure of PCF is kept secret. Our previous studies demonstrated that PCF was found to have an anti-apoptotic effect against UVB irradiation in HeLa cells[2] and hairless mice[3], which implied a potential clinical usage of preventing skin damage induced by UV light or solar light. However, relatively little is known about the effect of PCF on UVA plus UVB-mediated damage on normal human skin cells in vitro. HaCaT cell, a spontaneously immortalized human keratinocyte cell line, has similar characteristic to the normal keratinocyte line and thus may be considered as a good experimental model for testing the effects of PCF on UV irradiation-damaged skin cells.
UV irradiation can induce reactive oxygen species (ROS) accumulation and further cause cell apoptosis[4]. More and more studies have provided evidence that the activation of c-Jun amino-terminal kinase (JNK) is required for UV irradiation-induced apoptosis[5–7]. In addition, the JNK signaling pathway is known to play a fundamental role in UV-triggered, caspase-dependent apoptosis[8,9]. In view of this information, we hypothesized there may be a relation between ROS, JNK, and caspase-3 during the process of apoptosis induced by UV irradiation, and PCF may protect cells from apoptosis via blocking one of the signal pathways.
The purpose of this study was, by mimicking the action of environmental UV light on human skin, to investigate the anti-apoptotic function and protective mechanism of PCF on HaCaT cells damaged by UV irradiation.
Materials and methods
Reagents PCF (purity >96%) was purified and analyzed by high-performance liquid chromatography, dissolved in sterile deionized water, and stored at 4 °C. Antibodies against phospho-JNK, cleaved caspase-3 (17/19 kDa), β-actin, and Ac-DEVD-CHO (an inhibitor of caspase-3), were acquired from Cell Signaling (Beverly, MA, USA). 2',7'-Dichlorofluo-rescein diacetate (DCFH-DA), SP600125 (an inhibitor of JNK), and N-acetyl-L-cysteine (NAC, the scavenger of ROS) were obtained from Beyotime Biotechnology (Haimen, China).
Cell culture and treatment The HaCaT cells were cultured in the complete culture medium composed of Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA), 10% fetal bovine serum, 100 U/mL penicillin, and 100 U/mL streptomycin. The cells were maintained in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. UV irradiation was carried out using a UVA plus UVB light source (Beijing Normal University, Beijing, China). After standardization of conditions, the distance between the plate and the lamp was maintained at 7.0 cm, and the dose of UV irradiation was controlled by changing the time for radiation. When the confluent cells were radiating, the lid of the culture dish was removed and the culture medium was replaced by D-Hanks. The cells were randomly divided into several groups, including the control group (normal group), model group (UV-irradiated group), PCF group (PCF-pretreated and then UV-irradiated group), and the inhibitor groups (the corresponding inhibitor pretreated and then UV-irradiated group). PCF and the inhibitors of ROS, JNK, and caspase-3 were added into the medium for 2 h before radiation. After radiation, the cells were cultured in compete culture medium again for the appropriate time.
Cell viability assay Cell viability was evaluated via conventional 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) reduction assays. The HaCaT cells were planted into 96-well culture plates at an optimal density of 5×103 cells/mL with 100 µL culture medium per well. After 1–2 d culture to 90% confluence, the cells were radiated with different doses of UV irradiation, or incubated in medium containing different concentrations of PCF, or were pre-incubated with different concentrations of PCF for 2 h followed by UV irradiation. At 24 h after treatment, MTT solution was added to each well and incubated at 37 °C for 4 h. The medium was gently aspirated, and then 150 mL DMSO was added to each well to solubilize the formazan crystals. The absorbance of each sample was immediately measured using an ELISA reader at 490 nm.
Apoptosis assay Apoptosis in the HaCaT cells was analyzed by 2 methods: (i) Hoechst 33258 staining, where the cells were seeded on sterile cover glasses placed in the 6-well plates. At 18 h after treatment, the cells were fixed, washed twice with D-Hanks, and stained with Hoechst 33258 staining solution according to the manufacturer’s instructions (Beyotime Biotechnology, Jiangsu, China). Stained nuclei were observed under a fluorescence microscope; and (ii) DNA fragmentation, where the HaCaT cells were washed twice with D-Hanks solution and harvested. The cells were incubated in lysis buffer [150 mmol/L NaCl, 10 mmol/L Tris-HCl (pH 7.5), 10 mmol/L EDTA, 0.5% SDS, and 500 mg/L proteinase K] overnight at 50 °C. DNA was extracted with an equal volume of phenol and then extracted with an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) and precipitated with 0.1 volume of 3 mol/L NaAc and 2.0 volume of absolute ethanol, and incubated overnight at -20 °C. The DNA pellets were dissolved in Tris-EDTA buffer and analyzed on a 1.5 g/L agarose gel containing 0.5 g/L ethidium bromide, and visulized by UV transillumination.
Measurement of intracellular ROS levels Changes in the intracellular ROS levels were determined by measuring the oxidative conversion of cell permeable DCFH-DA to fluorescent dichlorofluorescein (DCF) by flow cytometry. After pretreatment with different concentrations of PCF or 5.68 mmol/L vitamin C for 2 h, the cells in the 6-well culture dishes were radiated by 4 J/cm2 UVA plus 10 mJ/cm2 UVB and cultured for another 30 min. Then the cells were collected and adjusted to 1×107/mL and incubated with DCFH-DA at 37 °C for 20 min. Subsequently, the cells were washed 3 times with D-Hanks, and a cover was placed on top of the dish. The fluorescent signal intensity of DCF was detected by flow cytometry at an excitation wavelength of 488 nm and at an emission wavelength of 535 nm.
Detection of superoxide dismutase (SOD) and glutathione peroxidase (GSH-px) activities and total anti-oxidative capacity (T-AOC) The cells in the 6-well culture dishes were washed 3 times with ice-cold D-Hanks and lysed in the extraction buffer [50 mmol/L Tris-HCl (pH7.4), 1 mmol/L ethyleneglycol-bis(2-aminoethylether)tetraacetic acid, 150 mmol/L NaCl, 1% (v/v) Triton X-100, 1 mmol/L phenyl-methylsulfonyl, 10 µg/mL aprotinin, 10 mmol/L EDTA, 1 mmol/L NaF, and 1 mmol/L Na3VO4] on ice for 30 min. Then the cells were scraped from the plates and the lysates were subjected to 20 000×g centrifugation at 4 °C for 10 min. The amount of proteins in the cleared lysates was quantified with a bicincho-ninic acid assay (Beyotime Biotechnology, China). After determining the amount of total proteins in the supernatants, we detected T-AOC and the enzymes that include SOD and GSH-px using biochemical methods following the instructions for the reagent kits (Nanjing Institute of Jiancheng Biological Engineering, Nanjing, China).
Western blotting The phosphorylated JNK (P-JNK), JNK, and cleaved caspase-3 were analyzed by Western blotting. At 6 h (for P-JNK and JNK) or 9 h (for cleaved caspase-3) after treatment, the protein was extracted and quantitated as previously described in this study. An equal amount of protein was separated on a 10% SDS-PAGE and transferred electrophoretically to the nitrocellulose membranes. The membrane was blocked with 5% bovine serum albumin and 0.1% Tween-20 in Tris-buffered saline for 2 h at room tempera-ture. The bolts were incubated with antibodies against P-JNK, JNK, cleaved caspase-3, or β-actin (dilution 1:1000) overnight at 4 °C, and then with peroxidase-conjugated secondary antibodies (dilution 1:2000) for 2 h at room tempera-ture. The bands were detected using the diamino-benzidine detection kit (Boster Biotechnology, Wuhan, China), and the result was analyzed by Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
Statistical analysis Data are expressed as mean±SD. Statistical analysis was performed with one-way ANONA, followed by the Bonferroni test using Origin7.5 (OriginLab Corporation, Northampton, MA, USA). The difference were considered significant if P<0.05.
Results
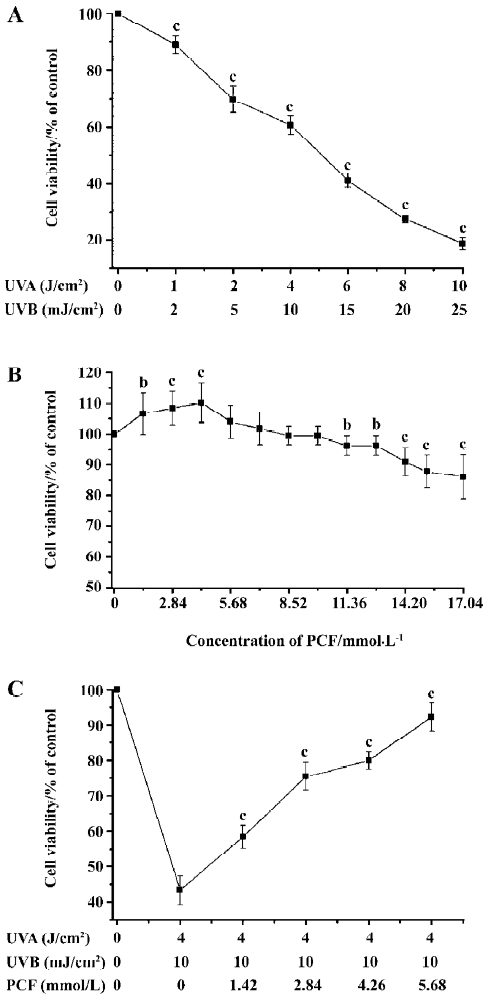
Effects of PCF and UV irradiation on the viability of HaCaT cells UV irradiation decreased cell viability in a dose-dependent manner. When the doses of UV irradiation were 4 J/cm2 UVA plus 10 mJ/cm2 UVB and 6 J/cm2 UVA plus 15 mJ/cm2 UVB, cell viability decreased to about 69% and 41% of the control group, respectively (Figure 1A). In this study, 4 J/cm2 UVA plus 10 mJ/cm2 UVB was applied to establish the apoptosis model. PCF treatment stimulated cell proliferation at low concentrations, but not at high concentration (Figure 1B). PCF pretreatment before radiation stimulated cell proliferation in a concentration-dependent manner in the range of 1.42–5.68 mmol/L (Figure 1C).
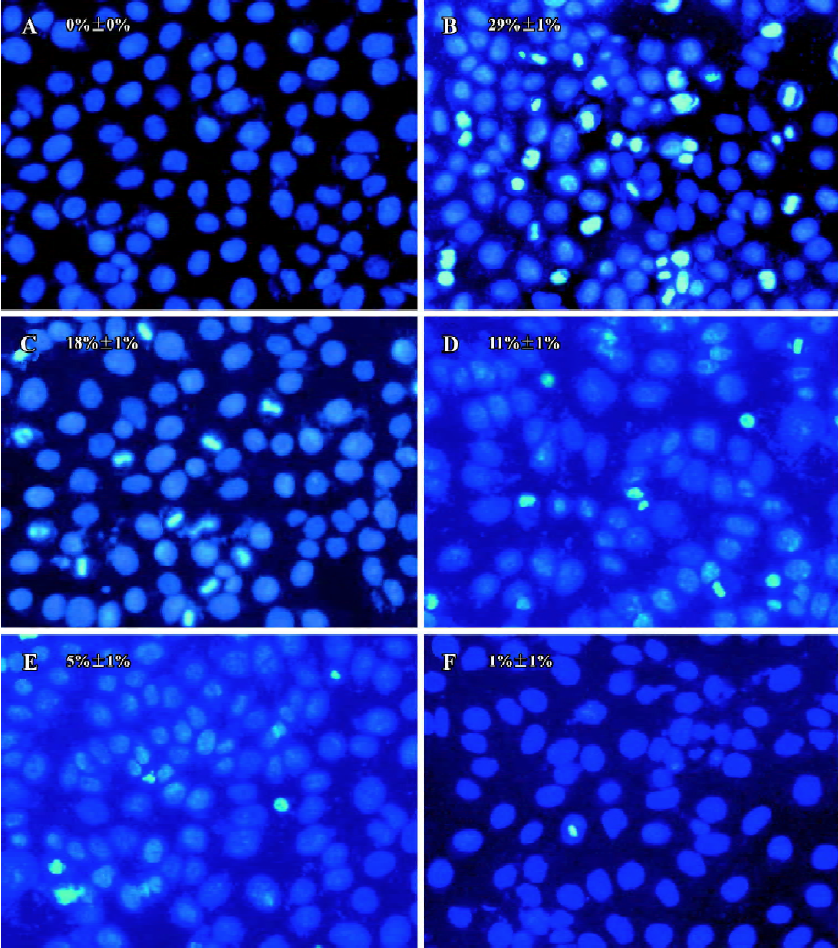
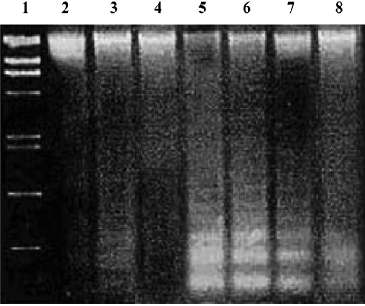
Effects of PCF on apoptosis of HaCaT cells induced by UV irradiation Hoechst 33258 staining of cells damaged by UV irradiation in the model group showed condensed, bright nuclei typical of apoptotic dead cells (Figure 2B), but the number of apoptotic nuclei in the PCF groups decreased in a concentration-dependent manner (Figure 2C, 2D, 2E), while almost no apoptotic nuclei were observed in the cells of the control and Ac-DEVD-CHO groups (Figure 2A, 2F). Since the cleavage of chromosomal DNA into fragments is a biochemical hallmark of apoptosis, the DNA fragmentation in HaCaT cells radiated by UV irradiation was examined by DNA laddering assay. With the cells in the control and Ac-DEVD-CHO groups, no obvious DNA fragmentation was observed, while clear DNA laddering was detected at 18 h after cell damage, and previous treatment by PCF prevented UV irradiation-induced DNA fragmentation in the HaCaT cells in a concentration-dependent manner (Figure 3).
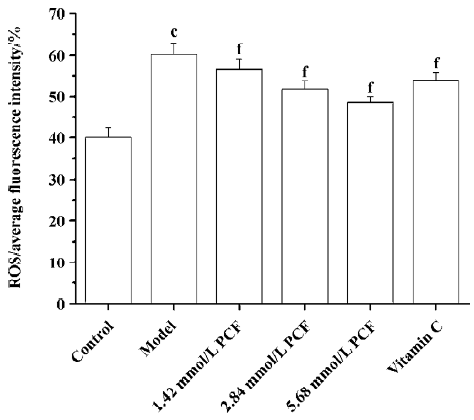
Effects of PCF on the intracellular ROS level in UV irradiation-treated HaCaT cells UV irradiation significantly increased the intracellular level of ROS. Pretreatment with PCF significantly inhibited the elevated intracellular concentration of ROS by UVA plus UVB in a concentration-dependent manner (Figure 4).
Effects of PCF on cellular SOD, GSH-px, and T-AOC UV irradiation can decrease the activities of SOD, GSH-px and T-AOC. Pretreatment with PCF for 2 h increased SOD, GSH-px, and T-AOC activities in a concentration-dependent manner (Table 1).
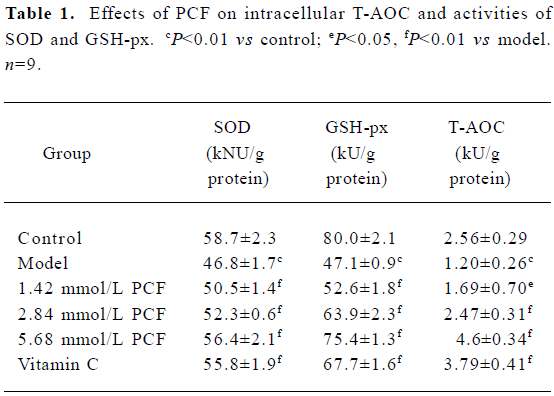
Full table
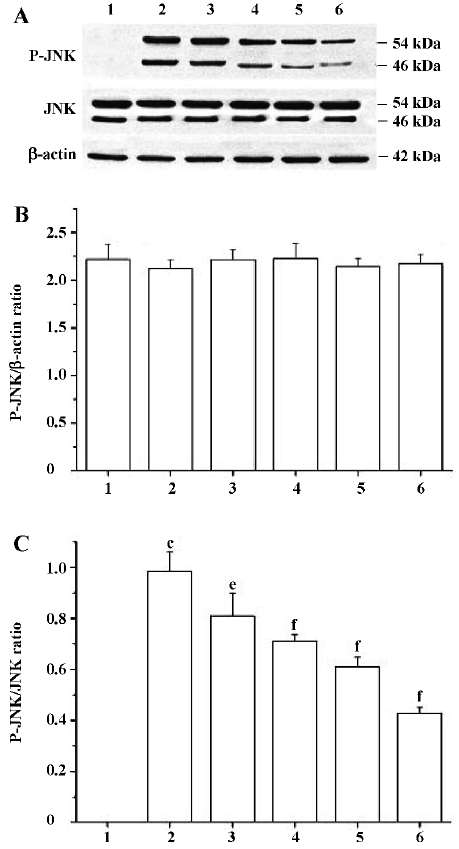
Effects of PCF on the UV irradiation-activated, JNK signaling pathway Western blotting studies showed that the total JNK protein level in cells at 6 h after UV irradiation was not significantly different from the cells of the control group. However, P-JNK protein expression was strongly increased. The treatment of cells with PCF inhibited UV irradiation-induced phosphorylation of JNK in a concentration-dependent manner. NAC, a scavenger of ROS, also decreased the P-JNK protein level in cells damaged by UV irradiation (Figure 5).
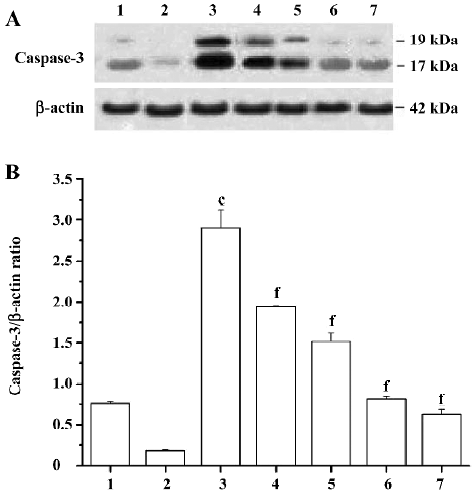
Effects of PCF on the expression of cleaved caspase-3 induced by UV irradiation During apoptosis, an inactive zymogen of precaspase-3 is cleaved into catalytically-active caspase-3 fragments, including a large fragment (17/19 kDa) and a small fragment (12 kDa). The expression of cleaved caspase-3 at 17/19 kDa in the HaCaT cells increased after exposure to UV irradiation; PCF pretreatment obviously decreased the expression of cleaved caspase-3 at 17/19 kDa in a concentration-dependent manner. Meanwhile, SP600125, an inhibitor of JNK, blocked the cleavage of precaspase-3 in the cells to a large extent. In addition, the decrease of cleaved caspase-3 at 17/19 kDa expression was found in the vitamin C group (Figure 6).
Discussion
Our results indicated that UVA plus UVB could inhibit the proliferation of HaCaT cells and induce cell apoptosis. So we successfully established the UV irradiation-induced apoptosis model of HaCaT cells by mimicking the action of environmental UV light on human skin in this study.
Apoptosis is a tightly regulated form of cell death and a multifactor-related process, including gene expression and mutation. The execution of the apoptosis program is characterized by morphological and biochemical changes. In our experiment, Hoechst 33258 staining and DNA laddering assay demonstrated that UV irradiation obviously induced the formulation of DNA laddering and karyopyknosis in cells, and PCF protected the cells from apoptosis. In the following studies, we tried to determine the mechanism of PCF preventing HaCaT cells from apoptosis induced by UVA plus UVB.
A role for oxidative stress in the induction of apoptosis is provided by studies where the addition of low levels of ROS can induce apoptosis, and the observation that various anti-oxidants such as N-acetylcysteine can inhibit cell death[10,11]. Additionally, ROS generation has been reported to occur following the treatment of cells with various agents, including UV irradiation and chemotherapeutic drugs[12]. Our results showed that the ROS level was low in the control group. Upon UV exposure, the ROS level increased, and PCF pretreatment reduced the ROS accumulation. Lee et al[13] suggested that UV irradiation could suppress the activities of anti-oxidative enzymes in cells. Several anti-oxidative enzymes, including GSH-px and SOD, scavenged free radicals produced by UV irradiation. In our experiments, PCF enhanced T-AOC and the activities of GSH-px and SOD. Generally speaking, the anti-oxidant system in healthy cells can automatically ameliorate insults induced by a slight increase of ROS. Our study showed that exposure to UVA plus UVB increased the intracellular ROS levels in cells by about 50% and decreased almost half of GSH-Px and T-AOC compared with normal cells. According to Rezvani et al’s[14] suggestion that UVB induced an increase in the ROS levels at 2 distinct stages: immediately following irradiation and around 3 h after irradiation, we speculated that the significant decrease of GSH-Px and T-AOC contributed to the amount of ROS in all stages. In the present study, we detected the ROS 30 min after UV radiation. Furthermore, with the exception of the formation of ROS, UV irradiation can induce apoptosis also via the triggering of death receptors or via DNA damage. Further study is needed to confirm our thoughts, but at least we can confirm that PCF works as a strong anti-oxidant.
The JNK signaling pathway is activated by a wide range of cellular stimulus such as UV light[15], radiation[16], cera-mide[17], DNA-damaging drugs[18], TNF-α[19], and interleukin 1[20]. In addition, mitogenic signals, including growth factors[21] and CD40 ligation[22] can induce the activation of JNK, which were originally identified through their involvement in c-Jun NH2-terminal phosphorylation following exposure to mammalian cells to short wavelength UV radiation[23]. The activation of JNK requires the phosphorylation of both Thr and Tyr residues located in its Thr-Pro-Tyr motif. In our experiments, the upregulation of P-JNK was observed at 6 h after the cells were exposed to UVA plus UVB, whereas, the increment of the ROS level was detected at 30 min after radiation. In addition, NAC, the scavenger of ROS, can block the activation of JNK. These results suggested that the ROS accumulation is upstream of the JNK activation induced by UV irradiation, which is consistent with the suggestion that inhibiting UVC light-induced ROS production inhibits JNK activation induced by UVC[24].
Our results also showed that HaCaT cell apoptosis by UVA plus UVB was dependent on the activation of caspase-3. Further studies showed that when SP600125, an inhibitor of JNK, was used, the activation of caspase-3 in the cells damaged by UV irradiation was blocked to a large extent. In addition, the upregulation of cleaved caspase-3 was found at 12 h after radiation, and was 6 h later than the activation of JNK. Although the activation of caspase-3 was not inhibited completely by SP600125, we can confirm that the activation of the JNK pathway led to the upregulation of the expres-sion of cleaved caspase-3 in HaCaT cells damaged by UVA plus UVB. Two possible mechanisms can explain how JNK activates caspase-3, one is the death receptor pathway. JNK activation may upregulate the expression of death receptor ligands[25]; the subsequent binding of FasL or TNF to its receptor induces the trimerization of the receptor and formation of a death-inducing complex. This complex recruits, via the adaptor molecule Fas-associated with death domain protein or TNF-R1-associated death domain, multiple procaspase-8 molecules, resulting in caspase-8 activation. The activated caspase-8 cleaves various proteins, including procaspase-3, which results in the activation of caspase cascades and the completion of apoptosis. Although it has been demonstrated that FasL can induce JNK activation, the activation is a delayed event[26] that probably requires the prior activation of caspases[27], which differs from this situation we describe. This places JNK activation in a primary role from where it may induce the expression of FasL or TNF to commit cells to apoptosis. The other mechanism that can explain how JNK activates caspase-3 is the mitochondrial death pathway. Bcl-2 family proteins localize or translocate to the mitochondrial membrane and modulate apoptosis by permeabilization of the inner and/or outer membrane. In vitro, JNK activation phosphorylates Bcl-2 and Bcl-XL[28–30], which significantly alters the susceptibility of cells to UV irradiation stimuli and causes cytochrome c release[31]. Then cytosolic cytochrome c forms an apoptosome complex with Apaf-1, dATP, and the initiator procaspase-9 to cause the activation of caspase-9 and trigger the subsequent effector caspase-3 activation[32–35], which results in the cleavage of cellular substrates and apoptosis. As to which pathway was involved in the activation of caspase-3 by JNK needs additional exploration.
In summary, our data demonstrated that the ROS-JNK-caspase-3-apoptosis cascade pathway is the main signal pathway of HaCaT cell apoptosis induced by UVA plus UVB. We found that PCF could protect HaCaT cells from damage by UV irradiation via scavenging ROS and increasing the activities of anti-oxidative enzymes as an anti-oxidant to block the signal pathway.
References
- Mu Y, Lv S, Ren X, Jin G, Liu J, Yan G, et al. UV-B induced kera-tinocyte apoptosis is blocked by 2-selenium-bridged β-cyclo-dextrin, a GPX mimic. J Photochem Photobiol B 2003;69:7-12.
- Yao RY, Wang CB. Protective effects of polypeptide from Chlamys farreri on HeLa cells damaged by ultraviolet A. Acta Pharmacol Sin 2002;23:1018-22.
- Wang CB, Yao RY, Liu ZT, Zhong WZ, Liu XP, Wang YJ. Protective effects of polypeptide from Chlamys farreri on hairless mice damaged by ultraviolet A. Acta Pharmacol Sin 2002;23:813-8.
- Petersen AB, Gniadecki R, Vicanova J, Thorn T, Wulf HC. Hydrogen peroxide is responsible for UVA-induced DNA damage measured by alkaline comet assay in HaCaT keratinocytes. J Photochem Photobiol B 2000;59:123-31.
- Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem 1996;271:31929-36.
- Katagiri C, Nakanishi J, Kadoya K, Hibino T. Serpin squamous cell carcinoma antigen inhibits UV-induced apoptosis via suppression of c-JUN NH2-terminal kinase. J Cell Biol 2006;172:983-90.
- Lo PK, Huang SZ, Chen HC, Wang FF. The prosurvival activity of p53 protects cells from UV-induced apoptosis by inhibiting c-Jun NH2-terminal kinase activity and mitochondrial death signaling. Cancer Res 2004;64:8736-45.
- Noguchi K, Kokubu A, Kitanaka C, Ichijo H. Kuchino. ASK1-signaling promotes c-Myc protein stability during apoptosis. Biochem Biophys Res Commun 2001;281:1313-20.
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000;288:870-4.
- Lennon SV, Martin SJ, Cotter TG. Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif 1991;24:203-14.
- Mcgowan AJ, Fernandes RS, Samali A, Cotter TG. Anti-oxidants and apoptosis. Biochem Soc Trans 1996;24:229-33.
- Gorman A, Mcgowan A, Cotter TG. Role of peroxide and superoxide anion during tumour cell apoptosis. FEBS Lett 1997;404:27-33.
- Lee J, Jiang S, Levine N, Watson RR. Carotenoid supplementation reduces erythema in human skin after simulated solar radiation exposure. Proc Soc Exp Biol Med 2000;223:170-4.
- Rezvani HR, Mazurier F, Cario-Andre M, Pain C, Ged C, Taieb A, et al. Protective effects of catalase overexpression on UVB-induced apoptosis in normal human keratinocytes. J Biol Chem 2006;281:17999-8007.
- Chen A, Davis BH. UV irradiation activates JNK and increases alpha (I) collagen gene expression in rat hepatic stellate cells. J Biol Chem 1999;274:158-64.
- Ruiter GA, Zerp SF, Bartelink H, van Blitterswijk WJ, Verheij M. Alkyl-lysophospholipids activate the SAPK/JNK pathway and enhance radiation-induced apoptosis. Cancer Res 1999;59:2457-63.
- Kurinna SM, Tsao CC, Nica AF, Jiffar T, Ruvolo PP. Ceramide promotes apoptosis in lung cancer-derived A549 cells by a mechanism involving c-Jun NH2-terminal kinase. Cancer Res 2004;64:7852-6.
- Hayakawa J, Depatie C, Ohmichi M, Mercola D. The activation of c-Jun NH2-terminal kinase (JNK) by DNA-damaging agents serves to promote drug resistance via activating transcription factor 2 (ATF2)-dependent enhanced DNA repair. J Biol Chem 2003;278:20582-92.
- Surapisitchat J, Hoefen RJ, Pi X, Yoshizumi M, Yan C, Berk BC. Fluid shear stress inhibits TNF-alpha activation of JNK but not ERK1/2 or p38 in human umbilical vein endothelial cells: Inhibitory crosstalk among MAPK family members. Proc Natl Acad Sci USA 2001;98:6476-81.
- Major CD, Wolf BA. Interleukin-1 beta stimulation of c-Jun NH2-terminal kinase activity in insulin-secreting cells: evidence for cytoplasmic restriction. Diabetes 2001;50:2721-8.
- Choi J, Park SY, Joo CK. Hepatocyte growth factor induces proliferation of lens epithelial cells through activation of ERK1/2 and JNK/SAPK. Invest Ophthalmol Vis Sci 2004;45:2696-704.
- Li YY, Baccam M, Waters SB, Pessin JE, Bishop GA, Koretzky GA. CD40 ligation results in protein kinase C-independent activation of ERK and JNK in resting murine splenic B cells. J Immunol 1996;157:1440-7.
- Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein-and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev 1993;7:2135-48.
- Charruyer A, Grazide S, Bezombes C, Muller S, Laurent G, Jaffrezou JP. UV-C light induces raft-associated acid sphingo-myelinase and JNK activation and translocation independently on a nuclear signal. J Biol Chem 2005;280:19196-204.
- Faris M, Kokot N, Latinis K, Kasibhatla S, Green DR, Koretzky GA. The c-Jun N-terminal kinase cascade plays a role in stress-induced apoptosis in Jurkat cells by up-regulating Fas ligand expres-sion. J Immunol 1998;160:134-44.
- Lenczowski JM, Dominguez L, Eder AM, King LB, Zacharchuk CM, Ashwell JD. Lack of a role for Jun kinase and AP-1 in Fas-induced apoptosis. Mol Cell Biol 1997;17:170-81.
- Cahill MA, Peter ME, Kischkel FC, Chinnaiyan AM, Dixit VM, Krammer PH, et al. CD95 (APO-1/Fas) induces activation of SAP kinases downstream of ICE-like proteases. Oncogene 1996;13:2087-96.
- Park J, Kim I, Young JO, Lee KW, Han PL, Choi EJ. Activation of c-Jun N-terminal kinase antagonizes an anti-apoptotic action of bcl-2. J Biol Chem 1997;272:16725-8.
- Fan M, Goodwin M, Vu T, Brantley-Finley C, Gaarde WA, Chambers TC. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J Biol Chem 2000;275:29980-5.
- Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Biol Cell 1999;19:8469-78.
- Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem 2000;275:3227.
- Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multi-meric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 1999;274:11549-56.
- Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol 2000;2:476-83.
- Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS. Caspase-9 can be activated without proteolytic processing. J Biol Chem 1999;274:8359-62.
- Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS. Dimer formation drives the activation of the cell death protease caspase 9. Proc Natl Acad Sci USA 2001;98:14250-5.