
Receptor subtype involved in α1-adrenergic receptor-mediated Ca2+ signaling in cardiomyocytes1
Introduction
The α1-adrenergic receptors (α1-AR) play a key role in the modulation of sympathetic nervous system activity as well as a site of action for therapeutic agents, such as antihypertensive drugs. Three subtypes of the α1-AR, α1A-AR, α1B-AR, and α1D-AR have been cloned and each has specific tissue distribution and pharmacological properties[1,2]. All subtypes of the α1-AR are Gq protein-coupled receptors, which upon activation, catalyze the cleavage of poly-phosphoinositide into dual signaling molecules, inositol 1,4,5-trisphosphate (IP3), and diacylglycerol via the activation of phospholipase C. IP3 leads to the opening of IP3 receptor channels at the endoplasmic/sarcoplasmic reticulum, and subsequently the release of intracellular Ca2+, while the activation of protein kinase C is the downstream signaling pathway for diacylglycerol[3,4]. Through these signal transduction pathways, the intracellular responses upon α1-AR stimulation are induced.
Accumulating studies have indicated that the α1-AR system appears to play a role in cardiac growth, cardiac contrac-tion, and cardiac automaticity in physiological condition[4–6], as well as in cardiac pathological processes, such as arrhythmo-genesis or cardiac adaptation to various situations[5,7,8]. Although the exact underlying mechanisms have not been conclusively determined, the increase in intracellular Ca2+ signaling, a common event seen in α1-AR stimulation, is considered to be a primary signaling pathway initiating acute as well as chronic cardiac function modulations by the α1-AR[2,7–11]. For instance, the α1-AR-mediated mobilization of Ca2+ from the sarcoplasmic reticulum contributes significantly to excitation–contraction coupling in atrial myocytes, and causes arrhythmogenic intracellular Ca2+ oscillations in the ischemic heart[7,9,10]. Additionally, α1-AR-mediated Ca2+ signaling is essential for the activation of calmodulin-dependent protein kinase II and nuclear factor of activated T cells, both of which signal a hypertrophic program of cardiac gene expression[8,12,13].
All 3 subtypes of α1-AR have been detected at the levels of messenger RNA as well as protein in the heart[14,15]. How-ever, the subtype of the receptor in the mediation of cardiac function is not clear. Many studies have suggested that the α1A- AR and α1B-AR appear to play major roles in the heart[3,6,15,16]. More recently however, the α1A-AR has been demonstrated to sufficiently induce cardiac arrhythmias and hypertrophy, while the α1B-AR seems less important[7,17,18]. Furthermore, the avtivation of α1B-AR even inhibits α1A-AR mediated cardiac remodeling[19], but plays a crucial role in the generation of dilated cardiomyopathy[16]. As an increase in intracellular Ca2+ is the primary signaling transduction pathway for α1-AR-mediated cardiac function[2,7–11], and the subtype involved is unclear, in this study we intended to identify the subtype of the α1-AR involved in mediating intracellular Ca2+ signaling by using neonatal rat ventricular myocytes (NRVM), which express all 3 α1-AR subtypes[14,15] and respond to α1-AR stimulation markedly in the profiles of intracellular Ca2+ signaling and hypertrophic growth[8,20,21].
Material and methods
Isolation and culture of cardiomyocytes NRVM were isolated from 1–2-d-old Sprague-Dawley rats by enzymatic digestion with 0.1% trypsin and 0.03% collegenase, as previously described[20]. Then the myocytes were plated onto laminin-coated, 35 mm dishes at a density of 0.5×103–0.8×103 cells/mm2 and cultured for 42 h in Dulbecco's modifi-ed Eagle’s medium (DMEM) and Medium 199 (4:1) containing 10% fetal bovine serum, 4 mmol/L L-glutamine, 100 units/mL penicillin/streptomycin, and 0.1 mmol/L 5-bromo-2-deoxyuridine to inhibit fibroblast proliferation. Before use, the myocytes were further cultured for 6 h in serum-free DMEM to eliminate any influence of some factors in the serum.
Confocal Ca2+ imaging The cultured NRVM were loaded with 4 µmol/L Fluo-4/AM (Molecular Probes, Eugene, OR, USA) at 37 °C for 30 min, and were then washed with 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid N-(2-Hydroxyethyl)piperazine-N'-(2-ethanesulfonic acid) (HEPES)- buffered salt solution (in mmol/L: NaCl 135, KCl 5, MgCl2 1, CaCl2 1.8, HEPES 10, and glucose 11, with pH 7.4 adjusted by NaOH) for 20 min. All the treatments for each dish were finished within 2 h.
Confocal images of fluo-4 fluorescence (excitation at 488 nm and emission detection at >515 nm) were obtained using a Leica SP2 inverted microscope equipped with a 63×, 1.3 numerical aperture, oil immersion objective. Time- lapsed (xy, 1.63 s/frame) or line-scan (xt, 2 ms/line, 0.15 µm/pixel) images were obtained with 1.5-µm axial resolution. Image data analysis used customer-devised routines coded in the Interactive Data Language Research System. All experiments were performed at room temperatures (22–24 °C).
Materials 5-Bromo-2-deoxyuridine, phenylephrine, 5-methylurapidil (5-Mu), chloroethylclonidine (CEC), BMY 7378 {8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro [4,5]decane-7,9-dione dihydrochloride}, propranolol, and prazosin were purchased from Sigma-Aldrich (St Louis, MO, USA). A61603 {N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetra hydronaphthalen-1-yl]methanesul-fonamide hydrobromide} was from Tocris (Ellisville, MO, USA).
Statistical analysis The data were analyzed and presented as mean±SEM. When appropriate, statistical comparison was carried out with 2-way paired or unpaired Student’s t-test or χ2 test. The accepted level of significance was P<0.05.
Results
In fluo-4-loaded NRVM, due to spontaneous action potentials as the trigger, rhythmic and spontaneous Ca2+ oscillations, were observed at a rate of 6.02±0.58 min-1 (n=12 experiments). Phenylephrine (PE, 10 µmol/L), a non-subtype specific agonist of the α1-AR[17,19,22], increases the frequency of the spontaneous Ca2+ transients (Figure 1A, upper panel), which was completely blocked with 1 µmol/L prazosin, an α1-AR antagonist, but not the β-AR antagonist propranolol at 1 µmol/L. This effect of PE is dose-dependent with an EC50 value (the concentration for inducing 50% of maximal response) of 2.3 µmol/L (Figure 1B). To determine the role of the α1-AR subtypes in [Ca2+]i regulation, we then examined the effects of subtype-specific antagonists on PE-mediated Ca2+ signal. As shown in Figure 1 (1A, bottom panel, 1C), pretreatment of myocytes with 5-Mu for 10 min, a specific inhibitor of the α1A-AR[23], caused dose-dependent suppression of the stimulatory response to PE (10 µmol/L) with an IC50 value (the concentration for 50% inhibition of agonist-induced response) of 6.7 nmol/L, and a complete abolishment was seen at a concentration of 30 nmol/L. In contrast, pretreatment of the cells for 30 min with CEC to inhibit the α1B-AR showed no influence, except that CEC at higher concentration (30 µmol/L) induced a 33.5% inhibition of PE-enhanced Ca2+ transients (Figure 1D). The alkylating agent CEC primarily inactivates the α1B-AR, but studies have shown that this compound can also produce partial inactivation of the other subtypes, especially α1A-AR, with prolonged exposure at high concentrations[24,25]. Thus, the partial blockade of the PE effect by CEC at higher concentration is most likely due to its non-specific inhibition of other subtypes. Nevertheless, blockade of the α1D-AR with BMY 7378 (0.1 µmol/L)[26,27] demonstrated no any influence in the PE effect (Figure 1D). Presently, the α1D-AR expressing much less than other subtypes in cardiomyocytes is functionally unknown in the heart[3,15]. These findings provide clues that the α1A-AR, not the α1B-AR and α1D-AR, may be the primary mediator of PE-regulated spontaneous Ca2+ transients.
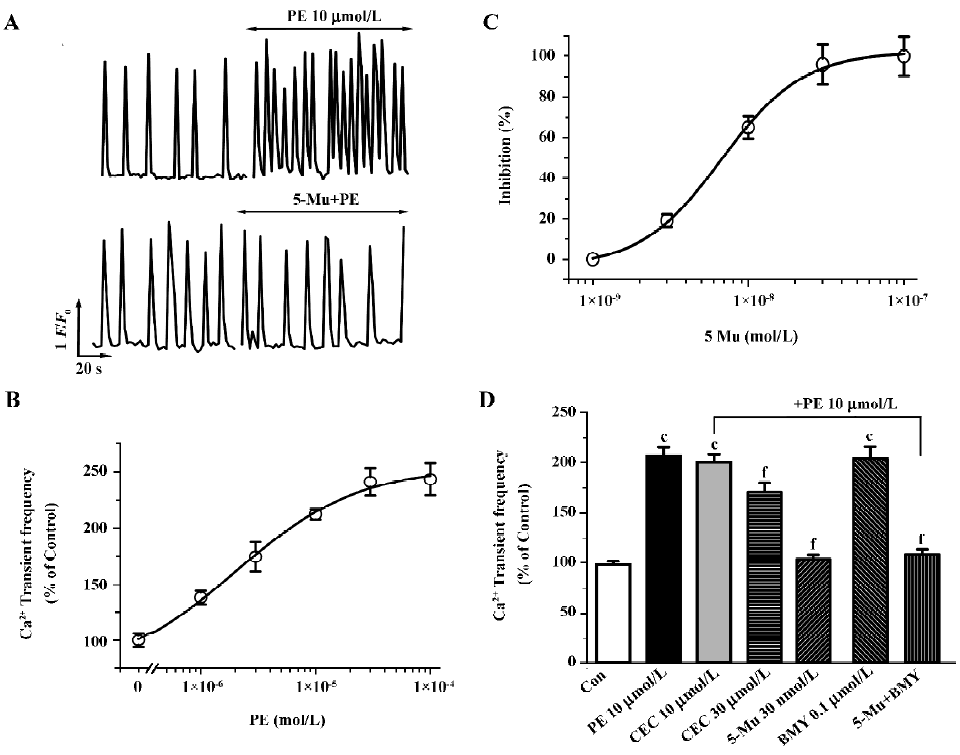
At present, the determination of the role of the α1A-AR versus the α1B-AR subtypes in mediating physiological responses to α1-adrenergic stimulation is difficult because of the paucity of highly selective antagonists specific for one subtype over the other. Therefore, to further discriminate the α1A-AR from other subtypes, we investigated subtype specific agonists in this protocol. So far no specific compound for the α1B-AR or the α1D-AR subtype is available, so we examined the effects of A61603, the recently described potent α1A-adrenergic agonist[28], and compared the dose-response characteristics of PE and A61603. As shown in Figure 2 (2A,2C), A61603 induced a dose-response increase in spontaneous Ca2+ transients with an EC50 value of 6.9 nmol/L, indicating a 330-fold greater potency for PE (2.3 µmol/L). Furthermore, we tried to find the concentration of A61603 at which stimulated Ca2+ transients with a similar potency to that of PE so as to evaluate and compare the inhibitory effect of 5-Mu. We observed that 30 nmol/L A61603 potentiated the rate of Ca2+ transients with an almost same potency as that of 10 µmol/L PE (2.11 and 2.09 times that of the control, respectively; Figure 2C,2D). Similar as its effect on PE (Figure 1D), 5-Mu (30 nmol/L), pretreatment of cells for 10 min, abolished the increment in Ca2+ transients induced by 30 nmol/L A61603 (Figure 2B,2D), while no effect was observed in 10 µmol/L CEC- or 0.1 µmol/L BMY 7378-treated cells (data not shown).
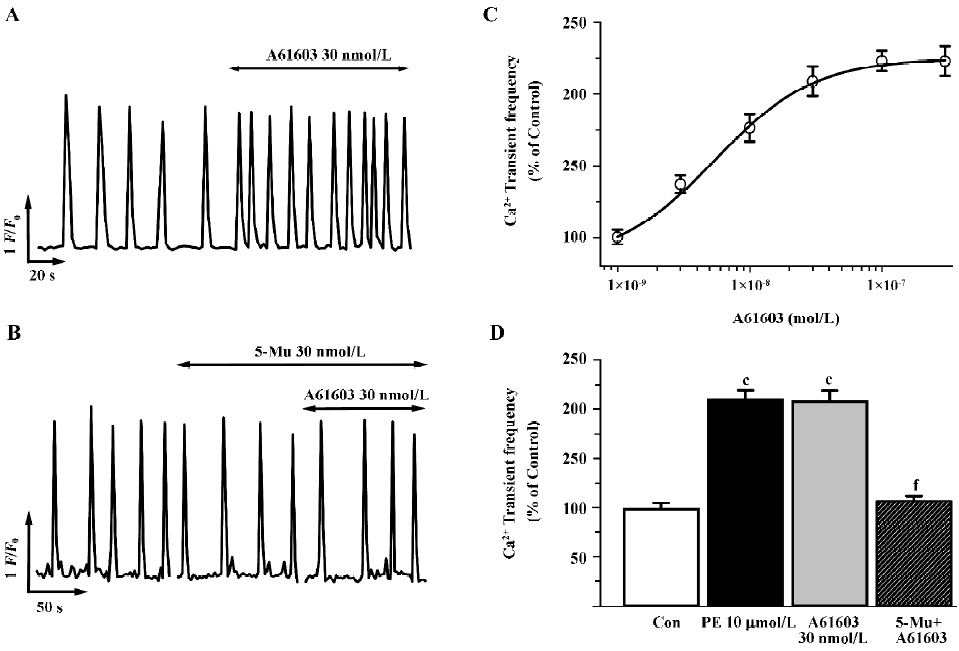
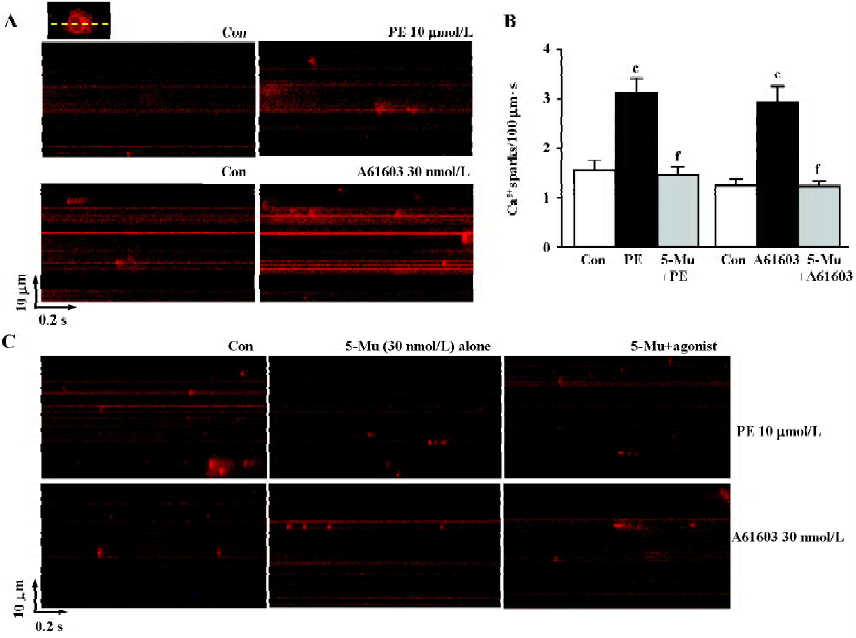
As spatial temporal Ca2+ sparks or waves constitute the elementary events of Ca2+ signaling in response to α1-adrenergic stimulation inside the cells, we then investigated the characteristics of Ca2+ sparks mediated by PE and A61603 in NRVM. With the aid of the line-scan confocal imaging of NRVM (Figure 3A, inset), we found that Ca2+ sparks occurred at a frequency of 1.56±0.2/100 µm·s or 1.27±0.15/100 µm·s in the control condition (in the absence of PE or A61603, respectively). PE (10 µmol/L) and A61603 (30 nmol/L) elicited a spark increase by 2.0- and 2.3-fold, respectively (Figure 3A, 3B), while the amplitude, width, and duration of the Ca2+ sparks were not altered by either treatment of the agonists. Consistent with the data in global Ca2+ transients, the local Ca2+ release responses to PE and A61603 could be abolished by 30 nmol/L 5-Mu (Figure 3B, 3C), but not by 10 µmol/L CEC or 0.1 µmol/L BMY 7378 treatment (data not shown). Therefore, the similar responses to A61603 and PE from local Ca2+ release to global Ca2+ transients, and a complete abolishment of both effects by specific α1A-AR antagonist, strongly suggest that the α1A-AR subtype plays a major role in the α1-AR-associated regulation of intracellular Ca2+ signaling in NRVM.
Discussion
Increases in intracellular Ca2+ signaling have been implicated to be an essential signal transduction event in the regulation of cardiac functions by α1-AR stimulation[2,7–11,20,21]. However, the subtype involved is not clear. The present study demonstrates that the stimulatory responses of spontaneous Ca2+ oscillations to α1-AR activation were greatly sensitive to and selectively abolished by the α1A-AR antagonist, but not by antagonism of the α1B-AR or the α1D-AR subtype (Figure 1). Additionally, A61603, the novel α1A-AR-selective agonist, exhibited a 330-fold greater potency than PE in stimulating spontaneous Ca2+ transient activity (Figure 2). This is in agreement with the findings that A61603 produces 340- and 330-fold greater potency than PE in stimulating sarcolemmal Na-H exchange activity in rat ventricular myocytes and in inducing contraction of the rat vas deferens, respectively. These physiological activities of α1-AR activation are further confirmed to be mediated by α1A-AR selectively[22,28]. Furthermore, Ca2+ sparks, an important event of the local Ca2+ releasing activity, were also stimulated by PE and A61603 with almost an equal potency with that in stimulating Ca2+ transients. This response to PE or A61603 is consistently abolished by the α1A-AR antagonist, but not by α1B-AR or α1D-AR inhibition (Figure 3). Taking these results together, these observations provide supportive evidence that α1-adrenergic stimulation of Ca2+ signaling activity is mediated selectively by the α1A-AR subtype.
Our previous study and those of others have shown that stimulated intracellular Ca2+ signaling plays an important role in the induction and perpetuation of cardiac hypertrophy by α1-AR activation[11,12,20,21]. Importantly, studies have shown that the α1A-AR subtype is sufficient in inducing hypertrophy in cultured cardiac myocytes[17,18] and is important in the development of the heart[6]. Thus, combined with the previous reports, the present study suggests that α1A-AR-mediated Ca2+ signaling response may assume greater significance in hypertrophy formation. Additionally, Ca2+ signal abnormity has also been suggested to play a key role in triggering ectopic automaticity in physiological as well as pathological circumstances, such as cardiac ischemia and heart failure[7,9,10] and α1-AR activation appears to be one of the important underlying mechanisms in this regard[7,29–31]. Interestingly, the α1A-AR has been suggested to be the crucial arrhythmogenic subtype by both in vivo and in vitro studies[7,9]. Therefore, taken these results together, the finding that the α1A-AR is the major subtype in intracellular Ca2+ signaling regulation during α1-AR activation may provide significant information for the functional roles of the α1-AR subtype and an alternate insight into the potential therapeutic candidates in heart remodeling and arrhythmias, particularly in humans as the α1A-AR appears to be the predominant subtype expressed in the human ventricular myocardium[32].
In summary, the present study has shown that the α1A-AR is the predominant subtype in regulating intracellular Ca2+ signaling for the α1-AR activation of neonatal rat ventricular myocytes, which may provide potential information for more specific drug development to hinder the cardiac remodeling process.
Acknowledgement
We thank Dr Qi-hua HE for outstanding technical assistance.
References
- Piascik MT, Perez DM. α1-Adrenergic receptors: new insights and directions. J Pharmacol Exp Ther 2001;298:403-10.
- Graham RM, Perez DM, Hwa J, Piascik MT. α1-Adrenergic receptor subtypes: molecular structure, function, and signaling. Circ Res 1996;78:737-49.
- Lazou A, Fuller SJ, Bogoyevitch MA, Orfali KA, Sugden PH. Characterization of stimulation of phosphoinositide hydrolysis by α1-adrenergic agonists in adult rat hearts. Am J Physiol 1994;267:H970-8.
- Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev 1999;51:651-90.
- Li K, He H, Li C, Sirois P, Rouleau JL. Myocardial alpha1-adrenoceptor: inotropic effect and physiologic and pathologic implications. Life Sci 1997;60:1305-18.
- O’Connell TD, Ishizaka S, Nakamura A, Swigart PM, Rodrigo MC, Simpson GL, et al. The α1A/C- and α1B-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J Clin Invest 2003;111:1783-91.
- Harrison SN, Autelitano DJ, Wang BH, Milano C, Du XJ, Woodcock EA. Reduced reperfusion-induced Ins(1,4,5)P3 generation and arrhythmias in hearts expressing constitutively active alpha1B-adrenergic receptors. Circ Res 1998;83:1232-40.
- Hunton DL, Lucchesi PA, Pang Y, Cheng X, Dell’Italia LJ, Marchase RB. Capacitative calcium entry contributes to nuclear factor of activated T cells nuclear translocation and hypertrophy in cardiomyocytes. J Biol Chem 2002;277:14266-73.
- Tolg R, Kurz T, Ungerer M, Schreieck J, Gorge B, Richardt G. Influence of alpha- and beta-adrenoceptor antagonists on ventricular fibrillation in ischemic rat hearts. Naunyn Schmiedebergs Arch Pharmacol 1997;356:62-8.
- Mackenzie L, Bootman MD, Laine M, Berridge MJ, Thuring J, Holmes A, et al. The role of inositol 1,4,5-trisphosphate receptors in Ca2+ signalling and the generation of arrhythmias in rat atrial myocytes. J Physiol 2002;541:395-409.
- Liu SJ, Kennedy RH. α1-Adrenergic activation of L-type calcium current in rat ventricular myocytes: perforated patch clamp recordings. Am J Physiol 1998;274:H2203-7.
- Sugden PH. Signaling in myocardial hypertrophy: life after calcineurin? Circ Res 1999;84:633-46.
- Frey N, McKinsey TA, Olson EN. Decoding calcium signals involved in cardiac growth and function. Nat Med 2000;6:1221-7.
- Luther HP, Podlowski S, Schulze W, Morwinski R, Buchwalow I, Baumann G, et al. Expression of alpha1-adrenergic receptor subtypes in heart cell culture. Mol Cell Biochem 2001;224:69-79.
- Stewart AF, Rokosh DG, Bailey BA, Karns LR, Chang KC, Long CS, et al. Cloning of the rat α1C-adrenergic receptor from cardiac myocytes: α1C, α1B, and α1D mRNAs are present in cardiac myocytes but not in cardiac fibroblasts. Circ Res 1994;75:796-802.
- Lemire I, Ducharme A, Tardif JC, Poulin F, Jones LR, Allen BG, et al. Cardiac-directed overexpression of wild-type alpha1B-adrenergic receptor induces dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 2001;281:H931-8.
- Autelitano DJ, Woodcock EA. Selective activation of alpha1A-adrenergic receptors in neonatal cardiac myocytes is sufficient to cause hypertrophy and differential regulation of alpha1-adrenergic receptor subtype mRNAs. J Mol Cell Cardiol 1998;30:1515-23.
- Pönicke K, Schlüter KD, Heinroth-Hoffmann I, Seyfarth T, Goldberg M, Osten B, et al. Noradrenaline-induced increase in protein synthesis in adult rat cardiomyocytes: involvement of only α1-adrenoceptors. Naunyn-Schmiedebergs Arch Pharmacol 2001;364:444-53.
- Deng XF, Sculptoreanu A, Mulay S, Peri KG, Li JF, Zheng WH, et al. Crosstalk between alpha-1A and alpha-1B adrenoceptors in neonatal rat myocardium: implications in cardiac hypertrophy. J Pharmacol Exp Ther 1998;286:489-96.
- Luo DL, Gao J, Lan XM, Wang G, Wei S, Xiao RP, et al. Role of inositol 1,4,5-trisphosphate receptors in α1-adrenergic receptor-induced cardiomyocyte hypertrophy. Acta Pharmacol Sin 2006;27:895-900.
- Garcia KD, Shah T, Garcia J. Immunolocalization of type 2 inositol 1,4,5-trisphosphate receptors in cardiac myocytes from newborn mice. Am J Physiol Cell Physiol 2004;287:C1048-57.
- Yokoyama H, Yasutake M, Avkiran M. α1-Adrenergic stimulation of sarcolemmal Na+-H+ exchanger activity in rat ventricular myocytes: evidence for selective mediation by the a1A-adreno-ceptor subtype. Circ Res 1998;82:1078-85.
- Li HT, Long CS, Gray MO, Rokosh DG, Honbo NY, Karliner JS. Cross talk between angiotensin AT1 and alpha1-adrenergic receptors: angiotensin II downregulates alpha1a-adrenergic receptor subtype mRNA and density in neonatal rat cardiac myocytes. Circ Res 1997;81:396-403.
- Laz TM, Forray C, Smith KE, Bard JA, Vaysse PJ, Branchek TA, et al. The rat homologue of the bovine α1C-adrenergic receptor shows the pharmacological properties of the classical α1A subtype. Mol Pharmacol 1994;46:414-22.
- Perez DM, Piascik MT, Malik N, Gavin R, Graham RM. Cloning, expression, and tissue distribution of the rat homolog of the bovine α1C-adrenergic receptor provide evidence for its classification as the a1A subtype. Mol Pharmacol 1994;46:823-31.
- Deng XF, Chemtob S, Varma DR. Characterization of α1D-adreno-ceptor subtype in rat myocardium, aorta and other tissues. Br J Pharmacol 1996;119:269-76.
- Turnbull L, McCloskey DT, O’Connell TD, Simpson PC, Baker AJ. α1-Adrenergic receptor responses in α1AB-AR knockout mouse hearts suggest the presence of α1D-AR. Am J Physiol Heart Circ Physiol 2003;284:H1104-H9.
- Knepper SM, Buckner SA, Brune ME, DeBernardis JF, Meyer MD, Hancock AA. A-61603, a potent α1-adrenergic receptor agonist, selective for the α1A receptor subtype. J Pharmacol Exp Ther 1995;274:97-103.
- Kontula K, Laitinen PJ, Lehtonen A, Toivonen L, Viitasalo M, Swan H. Catecholaminergic polymorphic ventricular tachycardia: recent mechanistic insights. Cardiovasc Res 2005;67:378-87.
- Lanzafame AA, Turnbull L, Amiramahdi F, Arthur JF, Huynh H, Woodcock EA. Inositol phospholipids localized to caveolae in rat heart are regulated by alpha1-adrenergic receptors and by ischemia-reperfusion. Am J Physiol Heart Circ Physiol 2006;290:H2059-65.
- Ravingerova T, Pancza D, Ziegelhoffer A, Styk J. Preconditioning modulates susceptibility to ischemia-induced arrhythmias in the rat heart: the role of alpha-adrenergic stimulation and K(ATP) channels. Physiol Res 2002;51:109-19.
- Price DT, Lefkowitz RJ, Caron MG, Berkowitz D, Schwinn DA. Localization of mRNA for three distinct α1-adrenergic receptor subtypes in human tissues: implications for human α1-adrenergic physiology. Mol Pharmacol 1994;45:171-5.