
Effects of (-)-stepholidine on NMDA receptors: comparison with haloperidol and clozapine1
Introduction
Animal and imaging data support a novel model of schizophrenia that putative brain dopamine (DA) function imbalance, that is, the subcortical D2 receptor (D2R) hyperfunction and prefrontal cortical (PFC) D1 receptor (D1R) dysfunction, may be sequential to NMDA receptor (NMDAR) hypofunction in the PFC[1,2]. NMDAR are ligandgated ion channels and are composed of multiple subunits (NR1, NR2A-D, and NR3A-C)[3]. A remarkable property of NMDAR is its high permeability to the calcium ion (Ca2+). Several studies have shown that neuro-leptics may affect influence NMDAR directly or indirectly. Haloperidol (Hal), a typical neuroleptic, is a potent antagonist of D2R. The cyclic AMP (cAMP) pathway is enhanced by Hal via its D2R blockage and it can phosphorylate the NR1 subunit of the NMDAR, which is necessary for neuroleptic-mediated gene expression and may contribute to the therapeutic bene-fits as well as side effects of neuroleptics treatment[4]. Clozapine (Cloz), an atypical neuroleptic, is effective in treating positive and negative syndromes of schizophrenia and refractory schizophrenia. In contrast to Hal, Cloz treatment induces relatively few extrapyramidal side effects[5]. A number of neurotransmitters and receptors, including Glu, DA, could contribute to Cloz’s atypical profile and clinical efficacy[6]. Cloz, but not Hal, can facilitate Glu receptor-mediated neurotransmission in the PFC[7].
A direct interaction of neuroleptics with NMDAR has been demonstrated. Hal has been shown to inhibit NMDAR function in a subunit-selective manner with NR2B-containing receptors displaying more sensitivity to the drug than non-NR2B-containing receptors[8]. Cloz, on the other hand, inhibits both NR2A and NR2B-containing NMDAR[9]. It is noted that these effects of Hal and Cloz would be counteractive to their therapeutic effects since blockade of NMDAR exacerbates symptoms in schizophrenia[10].
(-)-Stepholidine (SPD) is a tetrahydroprotoberberine alkaloid isolated from the Chinese herb Stephania. Our previous studies have shown that SPD is an agonist at D1R, but acts as antagonist at D2R[2]. SPD exhibits a profile similar to that of atypical neuroleptics[2,11–13]. More recently, it has been shown that SPD possesses agonist action on D1R in the PFC pyramidal glutamic (Glu) neuron, by which it modulates firing activity of nucleus accumbens neurons[14]; it mimics the effect of Cloz, preferentially increasing c-fos expression in the PFC via DA and non-DA systems[15]. Moreover, it has been found that SPD (0.1–10 µmol/L) can potently enhance synaptically-evoked NMDA EPSC (excitatory postsynaptic currents) in PFC neurons recorded in brain slices[16]; SPD (50 µmol/L) also increased the amplitude under the similar brain slices experiments[17]. However, little is known about the effect of SPD on NMDA receptors by direct or indirect mechanisms at the molecular level.
This study is intended to elucidate whether SPD has a similar pharmacological property to NMDAR as Cloz or whether it is different from Cloz and Hal, using Ca2+ imaging techniques for studying NMDAR function. The calcium imaging technique has an advantage in studying NMDAR function without change of intracellular conditions, which is different from patch clamp recording. The latter might cause alterations in the concentrations of intracellular constituents, such as protein kinase or phosphatase (important constituents in modulating NMDAR activity). The direct effects of SPD were compared with Hal and Cloz on cloned NMDAR transiently expressed in HEK293 cells.
Materials and methods
Materials Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA), newborn calf serum (Si Ji Qing, Hangzhou, China), penicillin and streptomycin (Watson Biotechnologies, Shanghai, China), Fura-2 acetoxymethyl ester (Fura-2 AM, Molecular Probes, Eugene, OR, USA), pluronic-127 (Sigma, St Louis, MO, USA), l-Glu acid sodium and glycine (Gly, Sino-American Biotechnology, Beijing, China), and Hal and Cloz (RBI, Natick, MA, USA) were used in the experiments. (-)-Stepholidine was obtained from Shanghai Institute of Materia Medica (Chinese Academy of Sciences, Shanghai, China). (-)-Stepholidine and Hal were dissolved in DMSO at a final concentration of 100 mmol/L, and Cloz in 0.1 mol/L HCl at a final concentration of 100 mmol/L. All stock solutions were stored under -20 oC and diluted with extracellular solution immediately before use.
Cell culture and transfection The HEK293 cells, a generous gift from Dr Gang PEI (Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China), were maintained in DMEM supplemented with 10% newborn calf serum, 100 U/mL penicillin, and 100 U/mL streptomycin. The cells were plated at 2.5×104 onto acid-washed, poly-L-lysine coated glass coverslips (20 mm×20 mm). Transfections were performed when cell confluence reached about 80%. Rat NR1a, NR2A, or NR2B (generous gifts from Dr John WOODWARD, Medical University of South Carolina, Charleston, SC, USA), and enhanced green fluorescent protein (EGFP) plasmids were delivered at a ratio of 1:1:1 by the calcium-phosphated precipitation method with the modification of an additional 1 mmol/L ketamine to prevent excitotoxity during transfections[18]. After transfection and incubation for 24 h, the cells were used for the experiments. EGFP was used to identify the transfected cells.
Calcium imaging[19] The transfected cells were incubated with Fura-2 AM (4 μmol/L with 0.025% pluronic-127) for 45 min at 37 °C in extracellular solution (135 mmol/L NaCl, 5.4 mmol/L KCl, 1.8 mmol/L CaCl2, 10mmol/L glucose, and 5 mmol/L HEPES (2-[4-(2-Hydroxyethyl)-1-piperazinyl]ethanesulfonic acid); pH (6.8). After washing 3 times with Fura-free extracellular solution (pH 7.2), the dishes were mounted on the stage of an Olympus BX51WI upright microscope (Tokyo, Japan) and perfused continuously with modified Ringer’s solution at a flow rate of 1.2–1.5 mL/min. l-Glu/Gly were applied via a computer-controlled Y-tube (outer φ=100 µm) situated 80 µm above the cells. The cells were exposed to the alternating 340 nm and 380 nm light from a xenon lamp via DG4 (Sutter Instrument Company, Novato, CA, USA) every 2 s during and immediately after Glu/Gly application, and the images were acquired via a charge coupled device (CCD) camera (CoolSNAP HQ, Roper Scien-tific, Duluth, GA, USA). To minimize UV exposure, images were taken every 15–60 s between drug applications. Ratio images were generated using MetaFlour software from Universal Imaging (West Chester, PA, USA). Intracellular calcium concentrations ([Ca2+]i) were determined by the ratio of the intensity of the wavelengths at 340 and 380 nm. NMDA-dependent increases in [Ca2+]i were calculated by subtracting the average baseline value from the peak value obtained during Glu/Gly application. SPD, Hal, and Cloz were applied throughout the bath.
Data analysis Data were analyzed using Origin 6.0 (OriginLab Corporation, Northampton, MA, USA) and expressed as mean±SEM. The statistical significance of a drug’s effect was determined by comparing measures before and after drug injection using ANOVA followed by a post-hoc Tukey test[20].
Results
Calcium imaging study of NMDA receptor function To establish stable Ca2+ response induced by l-Glu/Gly, and to protect cells from potential damage due to UV exposure, [Ca2+]i induced by Glu/Gly was recorded at 2 s intervals and basal [Ca2+]i were recorded at 15–60 s intervals. Pseudocolour ratio images at wavelengths of 340 and 380 nm were taken (Figure 1A). Cells were locally perfused with Glu/Gly (100/10 µmol/L) for 3 s. An increase of [Ca2+]i was detected in cells expressing NR1a/NR2A or NR1a/NR2B receptors. The peak of [Ca2+]i was 3–7 s after Glu/Gly treatment and then returned to basal levels following washout for 300 s.
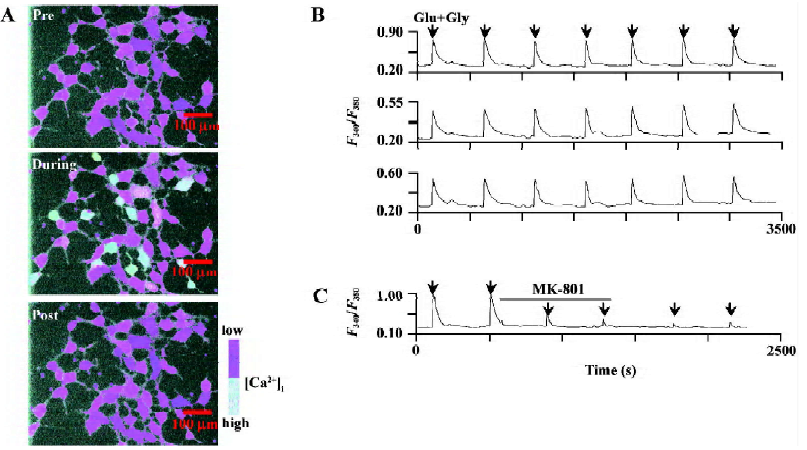
The local perfusion of Glu/Gly for 3 s could induce the reproducible increases in [Ca2+]i performed every 400 s duration (Figure 1B). Bath applications of (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine (MK-801) (1 µmol/L), a specific NMDAR antagonist, significantly blocked Glu/Gly-induced Ca2+ influx (80.3%±1.4% of inhibition, P<0.01, n=15; Figure 1C).
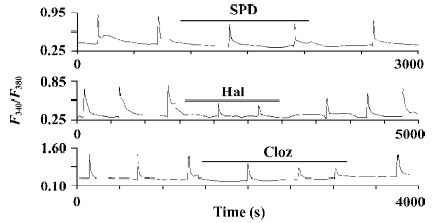
Effects of SPD, Hal, and Cloz on the NR2B subunit of NMDA receptors In the cells expressing the NR1a/NR2B receptor, both Hal and Cloz significantly inhibited the increase in [Ca2+]i induced by Glu/Gly. The inhibition induced by the 2 drugs could be observed at concentrations as low as 0.1–1 µmol/L (Figure 2). In contrast, SPD elicited only limited inhibition at a concentration of 100 µmol/L (18.1%±2.4% inhibition, P<0.01, n=26; Figures 2, 3). SPD produced no significant effect at less than 100 µmol/L (0.1 µmol/L, P=0.97, n=12; 1 µmol/L, P=0.52, n=7; and 10 µmol/L, P=0.15, n=10).
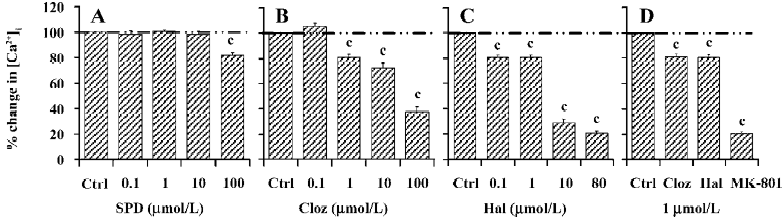
Although both Hal and Cloz inhibited the function of the NR1a/NR2B receptor, their effects were not entirely identified. Hal was much more potent than Cloz. Hal (0.1 µmol/L) inhibited Glu/Gly-induced Ca2+ influx (P<0.01, n=21), while Cloz produced no inhibition at the same concentration. However, when the concentration reached 10 µmol/L, Cloz inhibited the Glu/Gly-induced Ca2+ influx (Figure 3). The speed for Hal and Cloz operation also differed. Hal produced its peak effect at about 5 min after onset, while the maximal inhibition induced by Cloz required a 30 min perfusion. However, the effects of both Hal and Cloz were much less potent than that of MK-801 (Figure 2C).
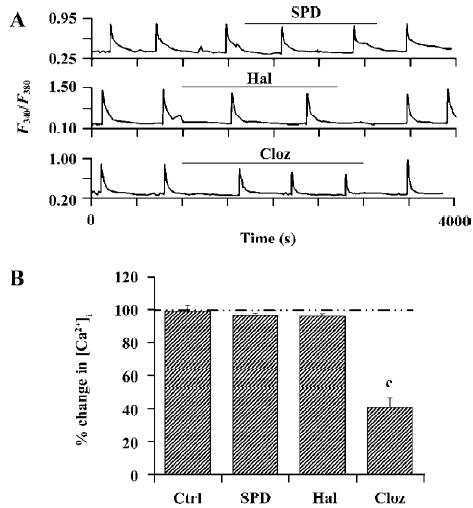
Effects of SPD, Hal, and Cloz on the NR2A subunit of NMDA receptors Cloz, not SPD (100 µmol/L) or Hal (50 µmol/L), significantly inhibited the function of the NR1a/NR2A receptor (Figure 4A, 4B). Cloz produced an average of 31.2%±4.2% (P<0.01, n=9) and 59.2%±5.8% (P<0.01, n=9) inhibition at 10 µmol/L and 100 µmol/L, respectively. A similar result was observed with the NR1a/NR2B receptors. Cloz also showed a slow onset in its effect on the NR1a/NR2A receptors (Figure 4A, lower section), which took 30 min to reach the maximum effect.
Discussion
The results of the present study found that SPD did not interact with the NR1a/NR2A or NR1a/NR2B receptors at low concentrations, unless 100 µmol/L SPD elicited certain inhibitory effects on NR1a/NR2B receptor-mediated Ca2+ influx. It appeared that SPD did not interact directly with NMDAR. In contrast with SPD, Hal and Cloz produced significant inhibitory effect on NMDAR-mediated Ca2+ influx. However, Hal selectively blocked the NR1a/NR2B receptors, whereas Cloz inhibited both NR1a/NR2B- and NR1a/NR2A-mediated responses.
In a previous study[8], Hal inhibited the function of NMDAR in a subunit-selective manner, with the NR2B-containing receptor more sensitive to the drug. However, Cloz inhibited Ca2+ influx mediated by both the NR2A and NR2B subunits of NMDAR[9]. Our results of Hal and Cloz were similar to the above mentioned both reports. Therefore, the different effects of the 3 drugs on NMDAR function may imply that SPD, Hal, and Cloz have different mechanisms in its antipsychotic use. In other words, Hal and Cloz have direct interaction with NMDAR, while SPD has an indirect one[21].
Our results showed that the significant inhibitory effect on Ca2+ influx by Hal was observed at 0.1 µmol/L, while Cloz was at 1 µmol/L. However, the clinic plasma concentra-tion of Hal and Cloz could reach 8–35 ng/mL (0.021–0.093 µmol/L)[22] and 350 ng/mL (1.07 µmol/L)[23,24], respectively, at which the 2 drugs may display their inhibitory effects on NMDAR-mediated Ca2+ influx. So what is the meaning of this inhibitory effect? There are two aspects with a contradiction. The inhibitory effect induced by Hal or Cloz is implicated to be beneficial for the negative symptoms of schizophrenia[25,26]. Alternatively, the inhibition of Hal and Cloz on NMDAR would limit their clinical therapeutic effect[27,28]. It may be suggested that the inhibition induced by Hal or the Cloz D2R blockade is the result of the inhibitory effect on the PFC NMDAR; subsequently, the inhibition would deteriorate the therapeutic effect, based on the DA function imbalance hypothesis[1,2].
As to the indirect interaction between SPD and NMDAR, we obtained definite evidence requiring postsynaptic density-95 (PSD-95)[21]. In an in vitro study, when PSD-95 was co-expressed with D1R and NMDAR in HEK293 cells, the activation of D1R could enhance NMDAR-mediated Ca2+ influx[21]. SPD hence could not display its enhancing effect on NMDAR under such experimental conditions without PSD-95. In the present study, SPD also could not affect NMDAR directly in this in vitro expression system for lack of D1R and other components.
On the other hand, in the experiment of brain PFC slices, including endogenic D1R and PSD-95, the patch clamp study recently demonstrated that SPD could increase the amplitude[17] or frequency[16] of sEPSC recording from the NMDAR of PFC pyramidal cells, respectively. Furthermore, the D1R agonist of SPD is mediated via intracellular protein kinase A and protein kinase C signaling pathways[16]. These results also strongly support that the D1R agonist of SPD enhances the NMDAR effect. Nevertheless, the enhancement of SPD on the NMDAR is obviously different from the inhibition elicited by Hal or Cloz on the NMDAR. According to these findings, 3 possible pathways have being proposed to explain how SPD can enhance NMDAR-mediated neurotransmission in the PFC. First, SPD possesses a unique D1R agonist effect[2], and the activation of D1R has been shown to enhance NMDAR-mediated responses in PFC neurons[16,29]. Second, SPD may act through the D1R to either enhance synaptic Glu release or to increase the response of the cell to NMDAR activation[16,17], as Cloz has been shown to increase spike-dependent presynaptic Glu release[30]. Third, SPD could result in a part effect through non-DA systems[15]. However, which class of non-DA receptors is responsible for the enhancing effect of SPD on NMDAR is still unknown.
Our results indicate that unlike Hal and Cloz, SPD did not directly affect the function of NMDAR at a low concentration. It is presumed that the modulatory effect of SPD on NMDAR function is an indirect effect via the D1R[16,21].
References
- Laruelle M, Kegeles LS, Abi-Dargham A. Glutamate, dopamine, and schizophrenia: from pathophysiology to treatment. Ann N Y Acad Sci 2003;1003:138-58.
- Jin GZ, Zhu ZT, Fu Y. (-)-Stepholidine: a potential novel antipsychotic drug with dual D1 receptor agonist and D2 receptor antagonist actions. Trends Pharmacol Sci 2002;23:4-7.
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev 1999;51:7-61.
- Leveque JC, Macias W, Rajadhyaksha A, Carlson RR, Barczak A, Kang S, et al. Intracellular modulation of NMDA receptor function by antipsychotic drugs. J Neurosci 2000;20:4011-20.
- Meltzer HY. The mechanism of action of novel antipsychotic drugs. Schizophr Bull 1991;17:263-87.
- Yamamoto BK, Cooperman MA. Differential effects of chronic antipsychotic drug treatment on extracellular glutamate and dopamine concentrations. J Neurosci 1994;14:4159-66.
- Arvanov VL, Wang RY. Clozapine, but not haloperidol, prevents the functional hyperactivity of N-methyl-D-aspartate receptors in rat cortical neurons induced by subchronic administration of phencyclidine. J Pharmacol Exp Ther 1999;289:1000-6.
- Ilyin VI, Whittemore ER, Guastella J, Weber E, Woodward RM. Subtype-selective inhibition of N-methyl-D-aspartate receptors by haloperidol. Mol Pharmacol 1996;50:1541-50.
- Levine JB, Martin G, Wilson A, Treistman SN. Clozapine inhibits isolated N-methyl-D-aspartate receptors expressed in xenopus oocytes in a subunit specific manner. Neurosci Lett 2003;346:125-8.
- Itil T, Keskiner A, Kiremitci N, Holden JM. Effect of phencyclidine in chronic schizophrenics. Can Psychiatr Assoc J 1967;12:209-12.
- Wu DC, Xing XZ, Wang WA, Xie H, Wang YH, Wang XY, et al. A double blind comparison trial of l-stepholidine and perphenazine in treatment of schizophrenia. Chin J New Drugs Clin Rem 2003;22:155-60. Chinese..
- Zhang XX, Sun BC, Jin GZ. Atypical neuroleptic properties of l-stepholidine. Sci China 1997;40:531-8.
- Ellenbroek BA, Zhang XX, Jin GZ. Effects of (-) stepholidine in animal models for schizophrenia. Acta Pharmacol Sin 2006;27:1111-8.
- Zhu ZT, Fu Y, Hu GY, Jin GZ. Modulation of medial prefrontal cortical D1 receptors on the excitatory firing of nucleus accumbens neurons elicited by (-)-stepholidine. Life Sci 2006;67:1265-74.
- Mo YQ, Jin XL, Chen YT, Jin GZ, Shi WX. Effects of l-stepholidine on forebrain Fos expression: comparison with clozapine and haloperidol. Neuropsychopharmacology 2005;30:261-7.
- Gao M, Liu CL, Yang S, Jin GZ. l-stepholidine increase the frequency of sEPSCs in rat prefrontal cortical neurons. Acta Pharmacol Sin 2007;28:627-33.
- Yang CR, Chen L. Targeting prefrontal cortical dopamine D1 and N-methyl-D-aspartate receptor interactions in schizophrenia treatment. Neuroscientist 2005;11:452-70.
- Cik M, Chazot PL, Stephenson FA. Optimal expression of cloned NMDAR1/NMDAR2A heteromeric glutamate receptors: a biochemical characterization. Biochem J 1993;296:877-83.
- Blevins T, Mirshahi T, Chandler LJ, Woodward JJ. Effects of acute and chronic ethanol exposure on heteromeric N-methyl-D-aspartate receptors expressed in HEK 293 cells. J Neurochem 1997;69:2345-54.
- Grishin AA, Gee CE, Gerber U, Benquet P. Differential calcium-dependent modulation of NMDA currents in CA1 and CA3 hippocampal pyramidal cells. J Neurosci 2004;24:350-5.
- Gu WH, Yang S, Shi WX, Zhen XC, Jin GZ. Requiring PSD-95 for dopamine D1 receptor modulating NMDA receptor function. Acta Pharmacol Sin 2007;28:756-62.
- Kelly MW, Perry PJ, Coryell WH, Miller DD, Arndt SV. Reduced haloperidol plasma concentration and clinical response in acute exacerbations of schizophrenia. Psychopharmacology (Berl) 1990;102:514-20.
- Bennett JA, Keck PE Jr. A target-dose finding study of clozapine in patients with schizophrenia. Ann Clin Psychiatry 1996;8:19-21.
- Perry PJ, Miller DD, Arndt SV, Cadoret RJ. Clozapine and norclozapine plasma concentrations and clinical response of treatment-refractory schizophrenic patients. Am J Psychiatry 1991;148:231-5.
- Goff DC, Tsai G, Levitt J, Amico E, Manoach D, Schoenfeld DA, et al. A placebo-controlled trial of D-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch Gen Psychiatry 1999;56:21-7.
- Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, Bar G, et al. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizo-phrenia. Biol Psychiatry 2005;57:577-85.
- Lane HY, Huang CL, Wu PL, Liu YC, Chang YC, Lin PY, et al. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to clozapine for the treatment of schizophrenia. Biol Psychiatry 2006;60:645-9.
- Jibson MD, Tandon R. New atypical antipsychotic medications. J Psychiatr Res 1998;32:215-28.
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci USA 2001;98:301-6.
- Chen L, Yang CR. Interaction of dopamine D1 and NMDA receptors mediates acute clozapine potentiation of glutamate EPSPs in rat prefrontal cortex. J Neurophysiol 2002;87:2324-36.